Figure S5. Extended ColocBoost xQTL analysis in aging human brain.#
ColocBoost was applied to 17 gene-level cis-xQTL datasets from the aging brain cortex of ROSMAP subjects.
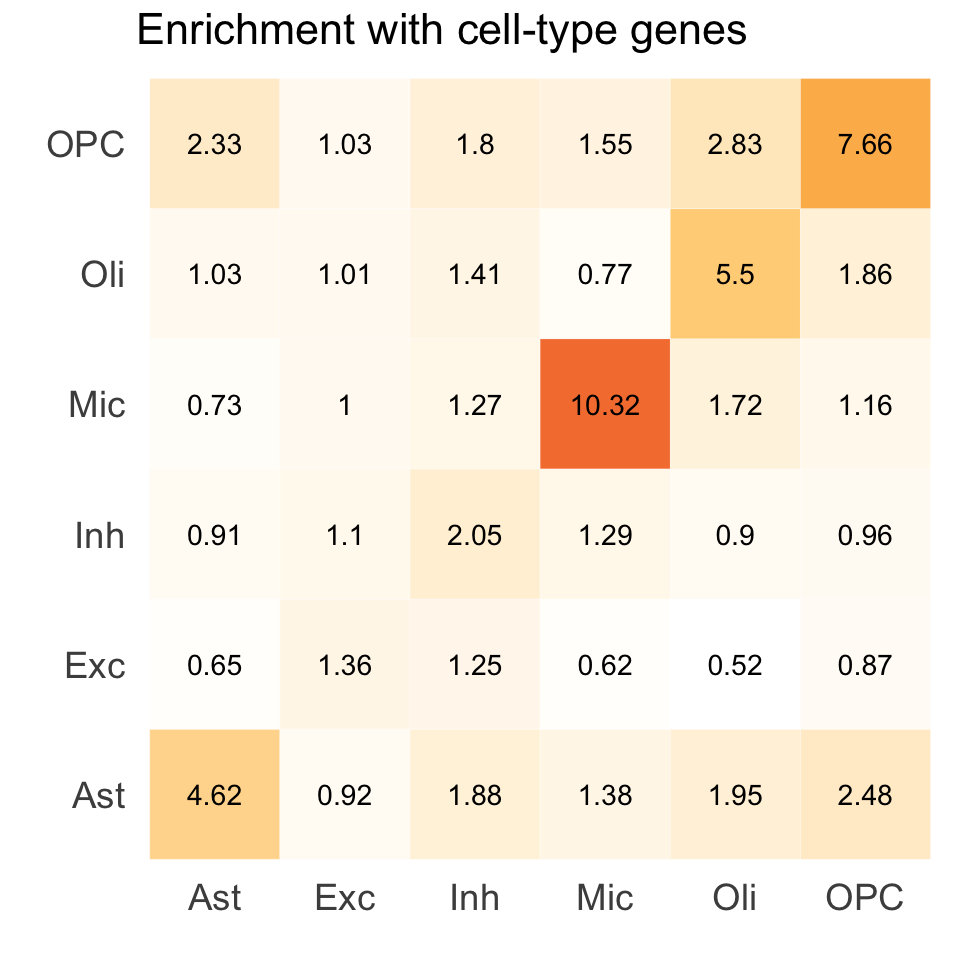
S5a. UpSet plot summarizing the top 80 colocalized events in xQTL-only ColocBoost analysis across 17 molecular traits.
S5b. UpSet plot displaying colocalization patterns across 3 molecular modalities (expression, splicing, and protein abundance) restricted to 3,655 genes with data available for all modalities.
S5c. The fraction of cell-type specific fine-mapping (measuring as 95% credible sets) for different brain cell types, that are recovered by cell-type specific colocalizations separately.
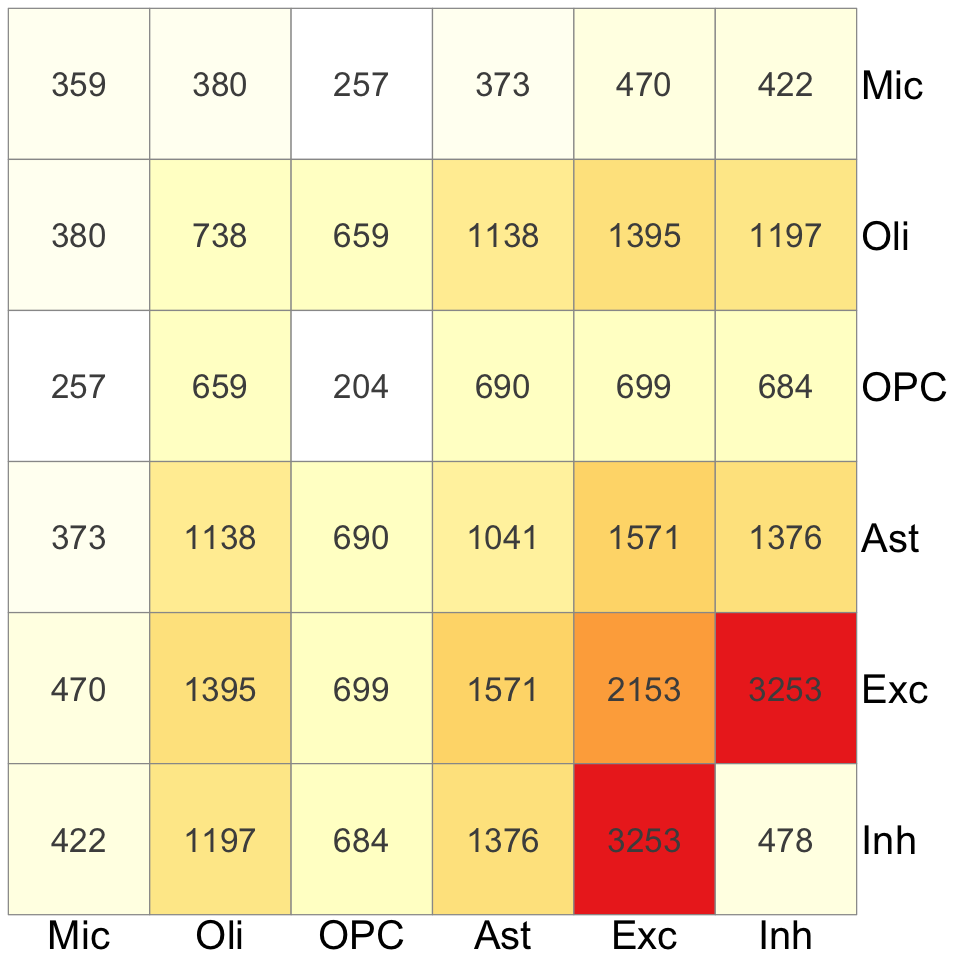
S5d. Summary of cell-type specific and cell-type shared colocalization events from (left penal) xQTL colocalization analysis spanning 17 molecular traits, and (right panel) eQTL colocalization analysis spanning eQTL data from 6 brain cell types and 4 bulk tissues. The diagonal entries indicate the number of cell-type specific colocalizations and off-diagonal entries indicate the number of colocalizations shared between each pairs of cell types.
S5e. Excess-of-overlap (EOO) analysis of the cell-type specific colocalization of eGenes with cell-type gene programs from external brain single-cell RNA-seq data.
Figure S5a#
UpSet plot summarizing the top 80 colocalized events in xQTL-only ColocBoost analysis across 17 molecular traits.
library("UpSetR")
res = readRDS("../../Main_Figures/Figure_3/data/xQTL_only_colocalization.rds")
all_pheno <- c("Mic","Ast","Oli","OPC","Exc","Inh","DLPFC","AC","PCC","Monocyte","pQTL",
"AC_productive","AC_unproductive","DLPFC_productive","DLPFC_unproductive","PCC_productive","PCC_unproductive")
coloc_pheno <- lapply(res$colocalized_phenotypes, function(cp){ strsplit(cp, "; ")[[1]] })
coloc <- lapply(all_pheno, function(y) {
pos <- sapply(coloc_pheno, function(cp) y %in% cp )
which(pos)
})
names(coloc) <- all_pheno
coloc_xQTL <- coloc[c(1:10, 14,15, 12,13, 16,17, 11)]
names(coloc_xQTL) <- c("Mic (DLPFC)", "Ast (DLPFC)", "Oli (DLPFC)", "OPC (DLPFC)", "Exc (DLPFC)", "Inh (DLPFC)",
"eQTL (DLPFC)", "eQTL (AC)", "eQTL (PCC)", "eQTL (Monocyte)",
"p-sQTL (DLPFC)", "u-sQTL (DLPFC)", "p-sQTL (AC)", "u-sQTL (AC)", "p-sQTL (PCC)", "u-sQTL (PCC)",
"pQTL (DLPFC)")
max_size <- max(sapply(coloc_xQTL, length))
p1 <- upset(fromList(coloc_xQTL),
order.by = "freq",
keep.order = T,
main.bar.color = "steelblue3",
sets.bar.color = "sandybrown",
text.scale = c(2.5,2,2.5,2,2,0), # Adjust font sizes for the main title, set names, set sizes, intersection sizes, and axis titles
matrix.color = "black", # Adjust the color of matrix dots
number.angles = 0, # Adjust the angle of number labels, useful for some plots
mb.ratio = c(0.5, 0.5), # Adjust the ratio of main bar and sets bar
point.size = 4, line.size = 1.5,
sets = c("Mic (DLPFC)", "OPC (DLPFC)", "Oli (DLPFC)", "Ast (DLPFC)", "Exc (DLPFC)", "Inh (DLPFC)",
"eQTL (DLPFC)", "eQTL (AC)", "eQTL (PCC)", "eQTL (Monocyte)",
"u-sQTL (DLPFC)", "p-sQTL (DLPFC)", "u-sQTL (AC)", "p-sQTL (AC)", "u-sQTL (PCC)", "p-sQTL (PCC)", "pQTL (DLPFC)"),
nsets = length(coloc_xQTL),
set_size.show = TRUE,
set_size.angles = 0,
set_size.numbers_size = 7,
set_size.scale_max = max_size + 0.55*max_size,
nintersects = 80,
mainbar.y.label = "Number of 95% CoS for\n each causal configuration",
sets.x.label = NULL)
options(repr.plot.width = 18, repr.plot.height = 10)
p1
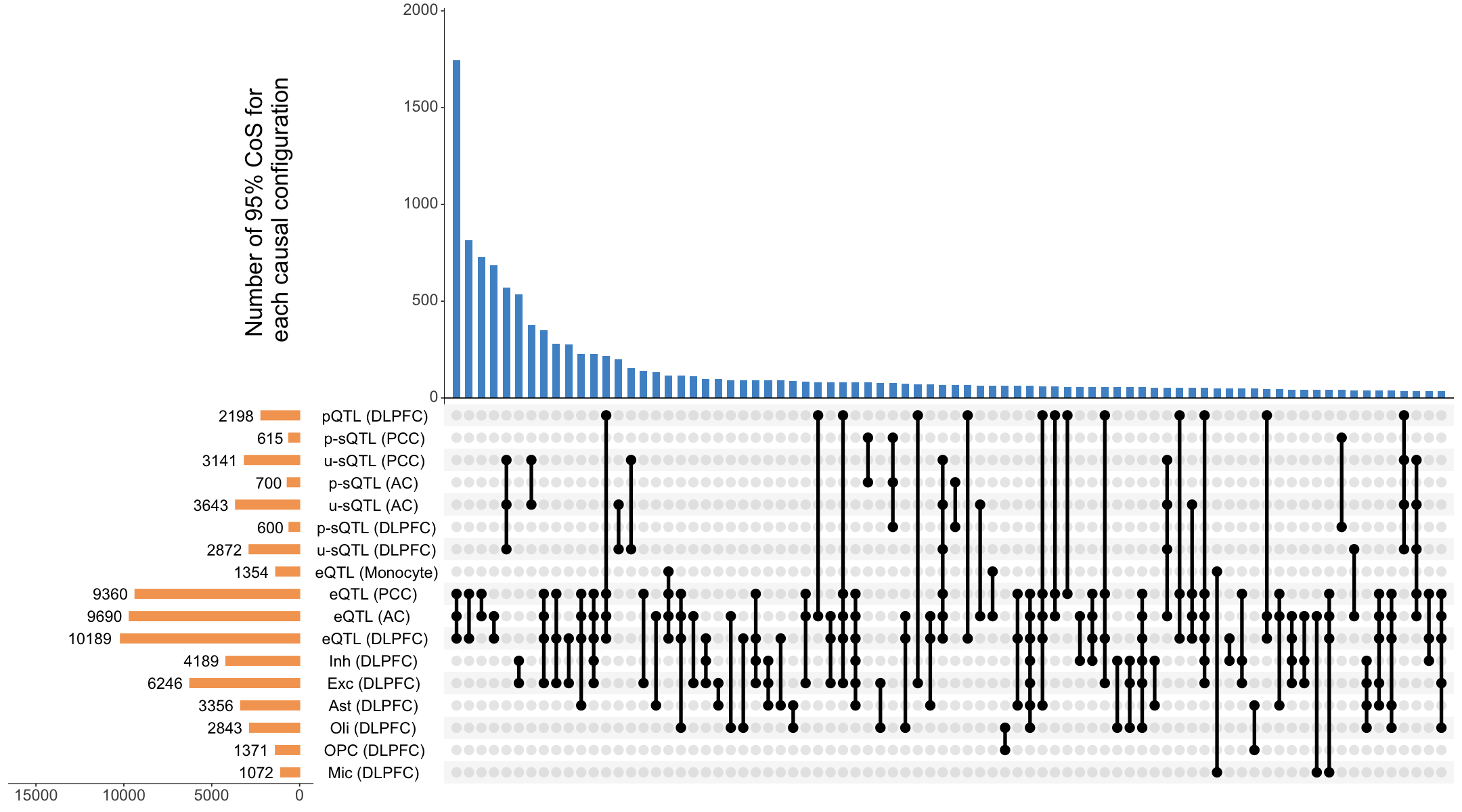
Figure S5b#
UpSet plot displaying colocalization patterns across 3 molecular modalities (expression, splicing, and protein abundance) restricted to 3,655 genes with data available for all modalities.
library("UpSetR")
res = readRDS("../../Main_Figures/Figure_3/data/xQTL_only_colocalization.rds")
genes <- read.table("Figure_S5b_three_xQTL_genes.txt", header = T)$x
library(tidyverse)
res <- res %>% filter(gene %in% genes)
all_pheno <- c("Mic","Ast","Oli","OPC","Exc","Inh","DLPFC","AC","PCC","Monocyte","pQTL",
"AC_productive","AC_unproductive","DLPFC_productive","DLPFC_unproductive","PCC_productive","PCC_unproductive")
coloc_pheno <- lapply(res$colocalized_phenotypes, function(cp){ strsplit(cp, "; ")[[1]] })
coloc <- lapply(all_pheno, function(y) {
pos <- sapply(coloc_pheno, function(cp) y %in% cp )
which(pos)
})
names(coloc) <- all_pheno
coloc_eQTL <- unique(unlist(coloc[1:10]))
coloc_pQTL <- coloc[[11]]
coloc_sQTL <- unique(unlist(coloc[12:17]))
coloc_xQTL <- list("eQTL" = coloc_eQTL,
"pQTL" = coloc_pQTL,
"sQTL" = coloc_sQTL)
names(coloc_xQTL) <- c(" eQTL\n (10 contexts)", " pQTL\n (1 context)", " sQTL\n (6 contexts)")
library("UpSetR")
library("ggplot2")
max_size <- max(sapply(coloc_xQTL, length))
p1 <- upset(fromList(coloc_xQTL),
order.by = "freq",
keep.order = T,
main.bar.color = "steelblue3",
sets.bar.color = "sandybrown",
text.scale = c(2,2,3,2,2,2.3), # Adjust font sizes for the main title, set names, set sizes, intersection sizes, and axis titles
matrix.color = "black", # Adjust the color of matrix dots
number.angles = 0, # Adjust the angle of number labels, useful for some plots
mb.ratio = c(0.5, 0.5), # Adjust the ratio of main bar and sets bar
point.size = 6, line.size = 1.5,
sets = c(" pQTL\n (1 context)", " sQTL\n (6 contexts)", " eQTL\n (10 contexts)"),
nsets = length(coloc_xQTL),
set_size.show = TRUE,
set_size.angles = 0,
set_size.numbers_size = 7,
set_size.scale_max = max_size + 0.55*max_size,
nintersects = 25,
mainbar.y.label = "Number of 95% CoS for\n each configuration",
sets.x.label = NULL)
options(repr.plot.width = 10, repr.plot.height = 6)
p1
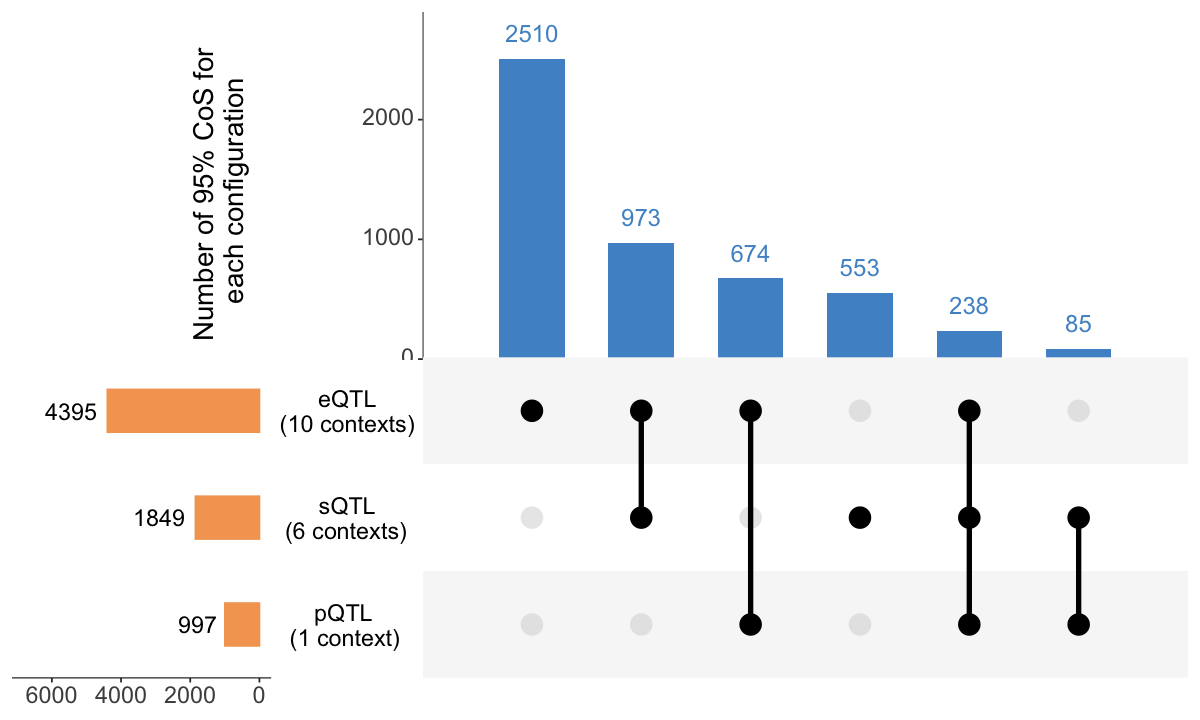
Figure S5c#
The fraction of cell-type specific fine-mapping (measuring as 95% credible sets) for different brain cell types, that are recovered by cell-type specific colocalizations separately.
library(ggsci)
data = readRDS("Figure_S5c.rds")
p <- ggplot(data, aes(y = CellType, x = Count, fill = Status)) +
geom_bar(stat = "identity", position = "stack") + # Stacked bar chart
scale_fill_npg() +
labs(
x = "Number of CoS",
y = NULL,
fill = "Proportion",
title = "Cell-type specific fine-mapping"
) +
theme_minimal(base_size = 15) +
theme(
plot.title = element_text( size = 28, hjust = 0.5 ),
axis.title.x = element_text( margin = margin(t = 0), size = 24), # Adjust x axis title margin
axis.title.y = element_text(margin = margin(r = 10), size = 24), # Adjust y axis title margin
axis.text.x = element_text(margin = margin(t = 0), size = 18), # Adjust x axis text margin
axis.text.y = element_text(margin = margin(r = 5), size = 18), # Adjust y axis text margin
legend.position = "inside",
legend.justification = c(0.95, 0.95),
legend.title = element_text(size = 0),
legend.text = element_text(size = 18),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.border = element_rect(color = "black", fill = NA, linewidth = 1.5)
)
options(repr.plot.width = 10, repr.plot.height = 6)
p
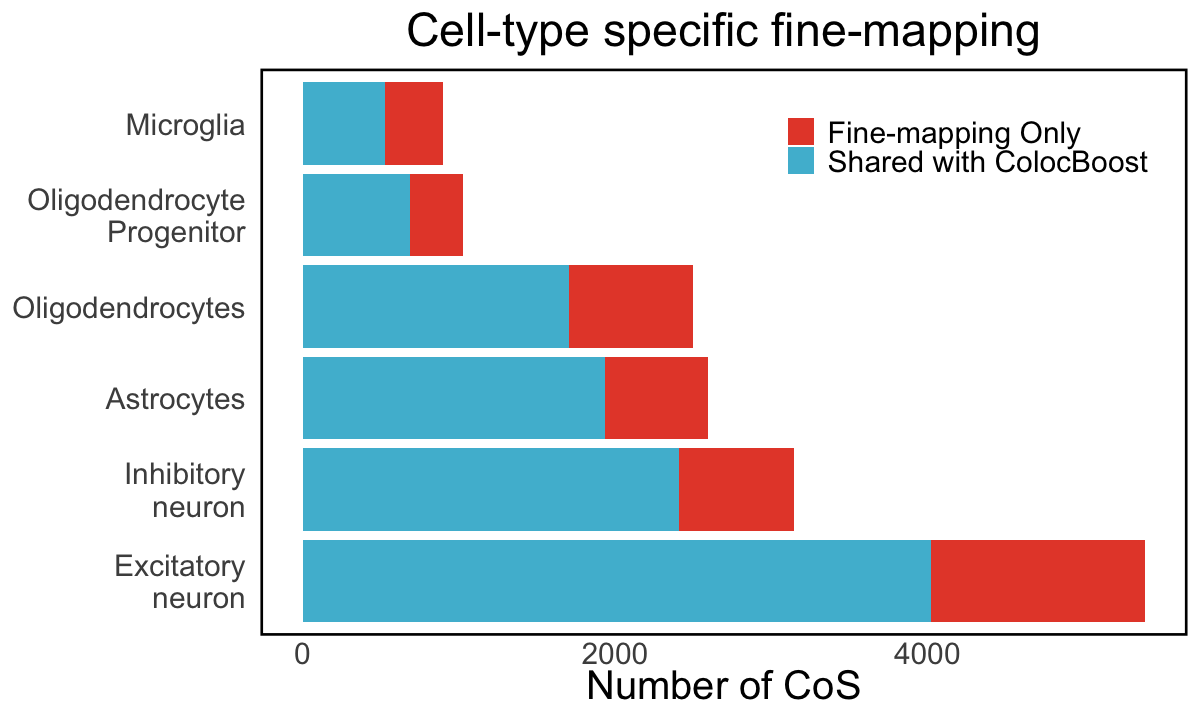
Figure S5d#
Summary of cell-type specific and cell-type shared colocalization events from (left penal) xQTL colocalization analysis spanning 17 molecular traits, and (right panel) eQTL colocalization analysis spanning eQTL data from 6 brain cell types and 4 bulk tissues. The diagonal entries indicate the number of cell-type specific colocalizations and off-diagonal entries indicate the number of colocalizations shared between each pairs of cell types.
options(repr.plot.width = 8, repr.plot.height = 8)
pair_xQTL = readRDS("Figure_S5d_xQTL.rds")
library(RColorBrewer)
color_palette <- colorRampPalette(c("white", brewer.pal(n = 9, name = 'YlOrRd')))(38)[1:28]
pheatmap::pheatmap(pair_xQTL, cluster_rows = FALSE, cluster_cols = FALSE,
display_numbers = TRUE, number_format = "%.0f",
fontsize = 16,
fontsize_row = 24,
fontsize_col = 24,
fontsize_number = 20,
angle_col = 0,
color = color_palette,
legend = FALSE
)
options(repr.plot.width = 8, repr.plot.height = 8)
pair_xQTL = readRDS("Figure_S5d_eQTL.rds")
library(RColorBrewer)
color_palette <- colorRampPalette(c("white", brewer.pal(n = 9, name = 'YlOrRd')))(38)[1:28]
pheatmap::pheatmap(pair_xQTL, cluster_rows = FALSE, cluster_cols = FALSE,
display_numbers = TRUE, number_format = "%.0f",
fontsize = 16,
fontsize_row = 24,
fontsize_col = 24,
fontsize_number = 20,
angle_col = 0,
color = color_palette,
legend = FALSE
)
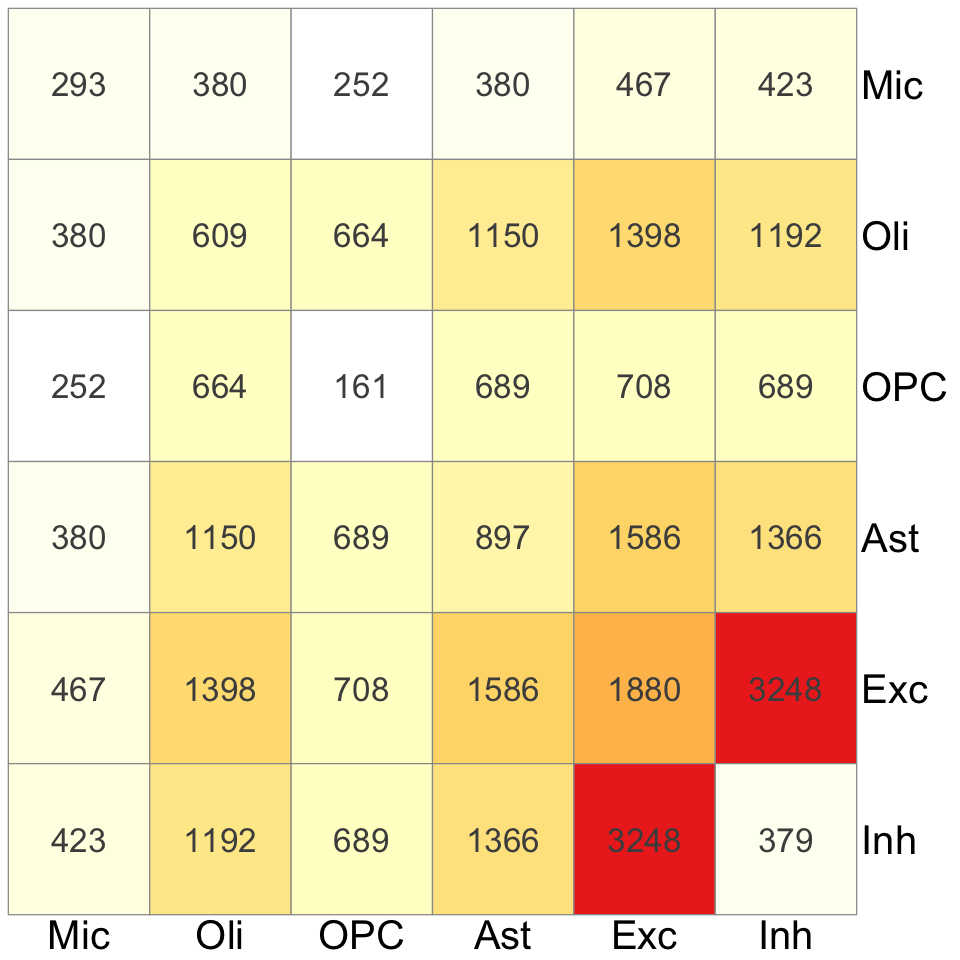
Figure S5e#
Excess-of-overlap (EOO) analysis of the cell-type specific colocalization of eGenes with cell-type gene programs from external brain single-cell RNA-seq data.
Enrich = readRDS("Figure_S5e.rds")
library(ggplot2)
library(reshape2)
corr_melt <- melt(Enrich)
p1 <- ggplot(corr_melt, aes(Var1, Var2, fill = value)) +
geom_tile(color = "white") +
scale_fill_gradient2(low = "white", high = "#E41C1C", mid = "#FEC661",
midpoint = 7, limit = c(0.5, 13), space = "Lab",
name="Enrichment") +
labs(title = "Enrichment with cell-type genes", x = "", y = "") +
theme_minimal(base_size = 15) +
theme(plot.title = element_text( size = 26 ),
axis.text.x = element_text(size = 22),
axis.text.y = element_text(size = 22),
panel.grid = element_blank(),
legend.position = "none") +
coord_fixed() +
geom_text(aes(Var1, Var2, label = round(value, 2)), color = "black", size = 6)
options(repr.plot.width = 8, repr.plot.height = 8)
p1