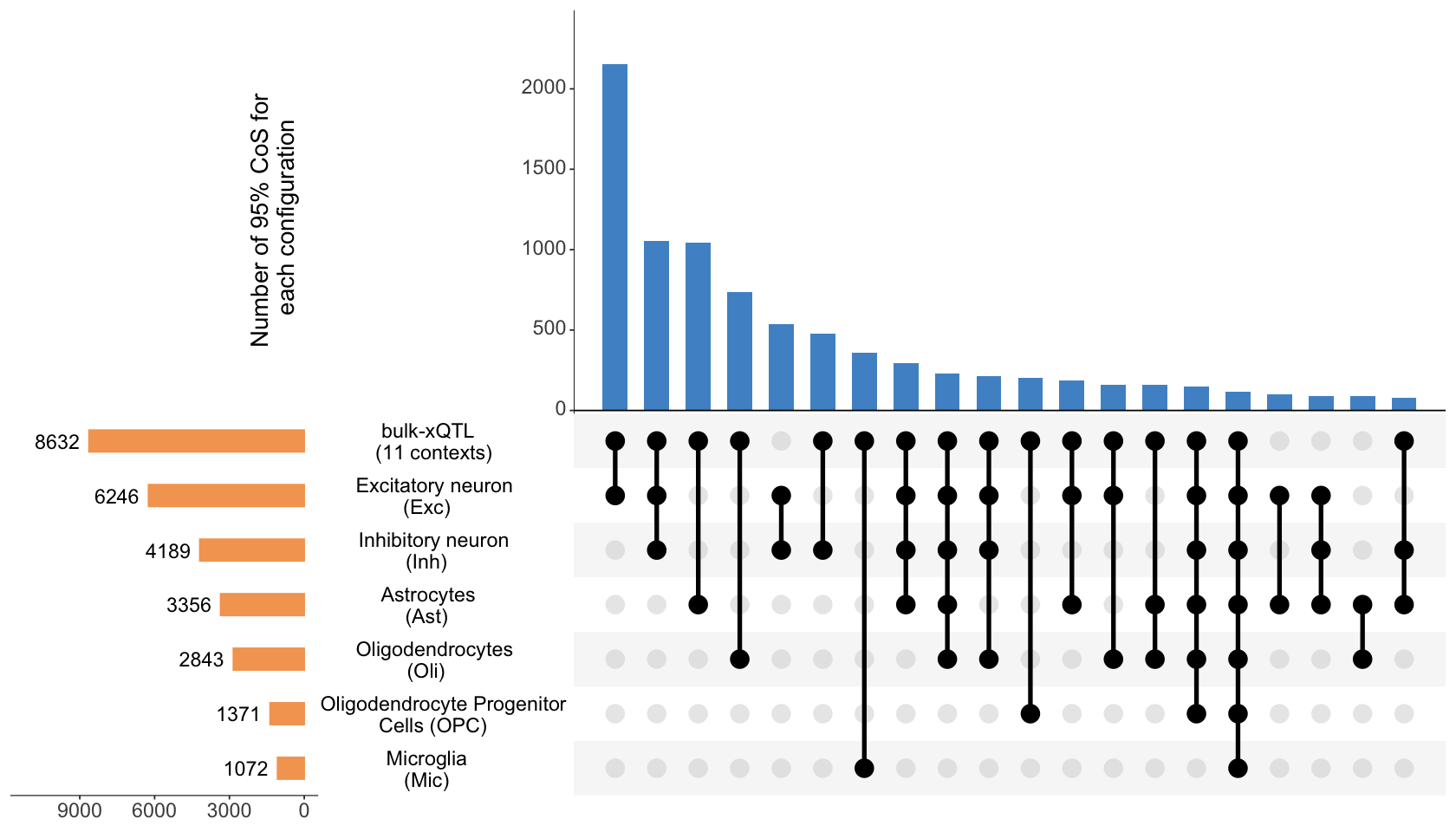
Figure 3c. UpSet plot summarizing the colocalization patterns across 6 pseudo-bulk brain cell-type eQTL data.#
UpSet plot summarizing the colocalization patterns across 6 pseudo-bulk brain cell-type eQTL data, along with bulk-xQTL data. We highlight putative causal variants with (i) shared effect across multiple brain cell types, and (ii) cell-type specific effect obtained through colocalization between bulk xQTL and pseudo-bulk eQTL for that cell type.
library(tidyverse)
library(ggpattern)
library(ggpubr)
library(cowplot)
res <- readRDS("data/xQTL_only_colocalization.rds")
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.5.1 ✔ tibble 3.2.1
✔ lubridate 1.9.4 ✔ tidyr 1.3.1
✔ purrr 1.0.4
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
Attaching package: ‘cowplot’
The following object is masked from ‘package:ggpubr’:
get_legend
The following object is masked from ‘package:lubridate’:
stamp
Organize input data#
all_pheno <- c("Mic","Ast","Oli","OPC","Exc","Inh","DLPFC","AC","PCC","Monocyte","pQTL",
"AC_productive","AC_unproductive","DLPFC_productive","DLPFC_unproductive","PCC_productive","PCC_unproductive")
coloc_pheno <- lapply(res$colocalized_phenotypes, function(cp){ strsplit(cp, "; ")[[1]] })
coloc <- lapply(all_pheno, function(y) {
pos <- sapply(coloc_pheno, function(cp) y %in% cp )
which(pos)
})
names(coloc) <- all_pheno
coloc_bulk_xQTL <- unique(unlist(coloc[7:17]))
coloc_pseudo <- coloc[1:6]
coloc_xQTL <- c(coloc_pseudo, list(coloc_bulk_xQTL))
names(coloc_xQTL) <- c("Microglia\n (Mic)", "Astrocytes\n (Ast)", "Oligodendrocytes\n (Oli)",
"Oligodendrocyte Progenitor\n Cells (OPC)", "Excitatory neuron\n (Exc)", "Inhibitory neuron\n (Inh)",
"bulk-xQTL\n (11 contexts)")
coloc_xQTL[[7]] <- intersect(coloc_xQTL[[7]], unlist(coloc_xQTL[1:6]))
UpSet plot#
library("UpSetR")
max_size <- max(sapply(coloc_xQTL, length))
p1 <- upset(fromList(coloc_xQTL),
order.by = "freq",
keep.order = T,
main.bar.color = "steelblue3",
sets.bar.color = "sandybrown",
text.scale = c(2,2,2.5,2,2,0), # Adjust font sizes for the main title, set names, set sizes, intersection sizes, and axis titles
matrix.color = "black", # Adjust the color of matrix dots
number.angles = 30, # Adjust the angle of number labels, useful for some plots
mb.ratio = c(0.5, 0.5), # Adjust the ratio of main bar and sets bar
point.size = 6, line.size = 1.5,
sets = c("Microglia\n (Mic)", "Oligodendrocyte Progenitor\n Cells (OPC)", "Oligodendrocytes\n (Oli)",
"Astrocytes\n (Ast)", "Inhibitory neuron\n (Inh)", "Excitatory neuron\n (Exc)",
"bulk-xQTL\n (11 contexts)"),
nsets = length(coloc_xQTL),
set_size.show = TRUE,
set_size.angles = 0,
set_size.numbers_size = 7,
set_size.scale_max = max_size + 0.3*max_size,
nintersects = 20,
mainbar.y.label = "Number of 95% CoS for\n each configuration",
sets.x.label = NULL)
options(repr.plot.width = 14, repr.plot.height = 8)
p1