GWAS / weaker signal simulation and ColocBoost#
Goal#
This notebook simulates the cases that the first trait is treated as GWAS, or any other traits that have a weaker signals than the other traits (eg. eQTL). The first trait will be designed to have a less heritability.
Input#
genofile: plink file of real genotyope, /mnt/vast/hpc/csg/FunGen_xQTL/ROSMAP/Genotype/plink_by_gene/extended_cis_before_winsorize_plink_files/*.bim
The other parameters can be found in simxQTL repo. https://github.com/StatFunGen/simxQTL.
Output#
An rds matrix, with genotype matrix X (dimension: m * n, m: number of sample, n: number of SNP ) and phenotype (trait) matrix (dimension: m * a, m : number of samples, a: number of simulated traits)
Example output:
result = readRDS("/home/hs3393/cb_Mar/simulation_data/simulation_GWAS/simulation_signal_0.02/causal_1/sample_1_h2g_0.02_GWAS_1.rds")
result$variant
- 824
- 824
# Check their p values?
library(susieR)
par(family = "sans")
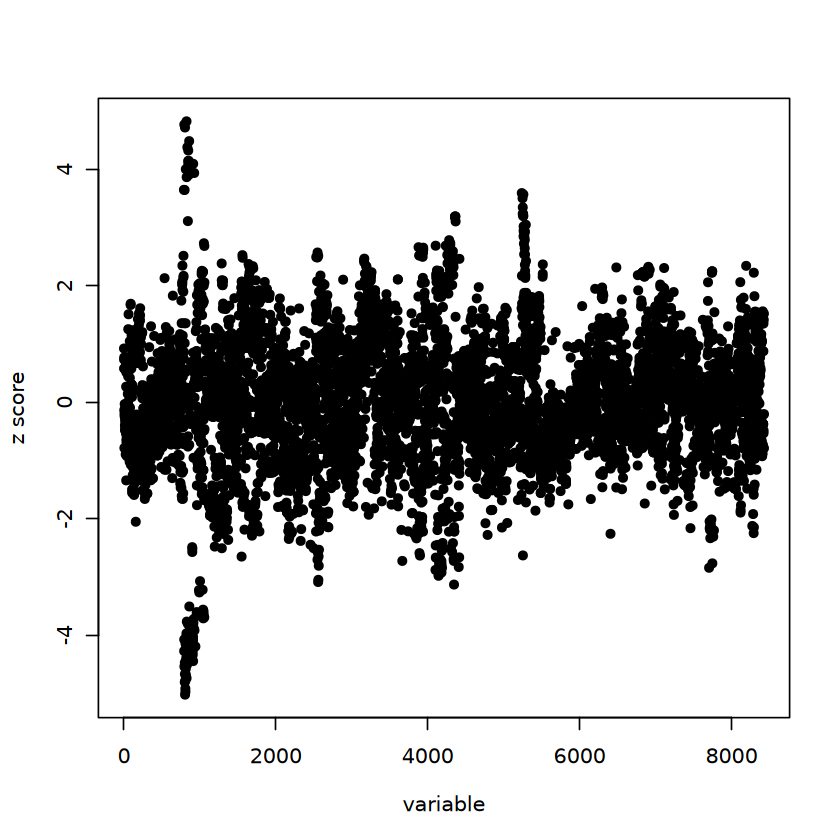
susie_res = susieR::susie(X = result$X, y = result$Y$Trait1, compute_univariate_zscore = TRUE)
susie_plot(susie_res, y = "z_original")
par(family = "sans")
susie_res = susieR::susie(X = result$X, y = result$Y$Trait2, compute_univariate_zscore = TRUE)
susie_plot(susie_res, y = "z_original")

In this case, the first trait (designed as GWAS) will not have fine-mapping CS result because of weak signal.
In contrast, the second trait have a CS result.
Phenotype simulation - 2 traits.#
Simulation code#
[simulation_GWAS]
parameter: genofile = paths
# pheno_file: give genotype file (in plink),we can read the gentype matrix. These files are separated by TADs.
parameter: cwd = path("output")
parameter: job_size = 30
parameter: walltime = "100h"
parameter: mem = "30G"
parameter: numThreads = 1
# for each variant, how many traits it randomly colocalize at
parameter: ntrait = 2
parameter: h2g = 0.05
parameter: ncausal = 1
parameter: share_pattern = "all"
# specify the number of traits (phenotypes)
parameter: container = ""
parameter: independent = False
input: genofile, group_by = 1
output: f'{cwd:a}/sample_{_index}_h2g_{h2g}_GWAS_{ncausal}.rds'
task: trunk_workers = 1, trunk_size = job_size, walltime = walltime, mem = mem, cores = numThreads, tags = f'{step_name}_{_output[0]:bn}'
R: expand = '${ }', stdout = f"{_output:n}.stdout", stderr = f"{_output:n}.stderr", container = container
library("MASS")
library("plink2R")
library("dplyr")
library("readr")
library("tidyverse")
# source some functions to read matrix and inpute the missing data
source("~/cloud_colocalization/simulation_code/simulate_linreg.R")
source("~/cloud_colocalization/simulation_code/misc.R")
# read the plink file
simu_file = ${_input:r}
geno <- read_plink(${_input:nr})
gene_name = str_extract(simu_file, "ENSG[0-9]+")
gene_tss_map = read_tsv("/home/hs3393/coloc/fungen-xqtl-analysis/resource/gene_cis_TADB_mapper.tsv")
# filter by distance with. TSS
TSS_pos = gene_tss_map$TSS[which(gene_tss_map$gene_id == gene_name)][1]
keep_index = which(geno$bim$V4 > TSS_pos - 1500000 | geno$bim$V4 < TSS_pos + 1500000)
geno$bed = geno$bed[,keep_index]
# filter out columns with missing rate > 0.1
imiss = 0.1
# filter out columns with MAF < 0.05
maf = 0.05
Xmat = filter_X(geno$bed, imiss, maf)
# To avoid that the matrix is too large, we use only the first 5000 SNPs in each TAD, if that region have more than 5,000
ncausal = ${ncausal}
ntrait = ${ntrait}
if (indep) {
LD_vars = 1 # Initialize LD_vars
if (ncausal == 1) {
# If only one causal variant, just sample it
vars = sample(1:ncol(Xmat), size = ncausal)
} else {
# Repeat sampling until selected variables are quasi independent
while (length(LD_vars != 0)) {
vars = sample(1:ncol(Xmat), size = ncausal)
cor_mat = cor(Xmat[, vars])
LD_vars = which(colSums(abs(cor_mat) > 0.3) > 1)
}
}
} else {
vars = sample(1:ncol(Xmat), size = ncausal)
}
B = matrix(0, nrow = ncol(Xmat), ncol = ntrait)
phenotype = list()
i = 1
beta = sim_beta_fix_variant(G = Xmat, causal_index = vars, is_h2g_total = FALSE)
B[, i] = beta
pheno_single = sim_multi_traits(G = Xmat, B = B[,i, drop = FALSE], h2g = ${h2g}, is_h2g_total = FALSE)
phenotype[[i]] = pheno_single$P
i = 2
beta = sim_beta_fix_variant(G = Xmat, causal_index = vars, is_h2g_total = FALSE)
B[, i] = beta
pheno_single = sim_multi_traits(G = Xmat, B = B[,i, drop = FALSE], h2g = 0.05, is_h2g_total = FALSE)
phenotype[[i]] = pheno_single$P
variant = list()
for(i in 1:ncol(B)){
variant[[i]] = which(B[,i] != 0)
}
X = Xmat
Y = bind_cols(phenotype)
colnames(Y) = paste0("Trait", c(1:ntrait))
data = list()
data[["X"]] = Xmat
data[["Y"]] = Y
data[["variant"]] = variant
saveRDS(data, ${_output:r})
Simulation GWAS - 5 traits#
[GWAS_5trait]
parameter: genofile = paths
# pheno_file: give genotype file (in plink),we can read the gentype matrix. These files are separated by TADs.
parameter: cwd = path("output")
parameter: job_size = 30
parameter: walltime = "100h"
parameter: mem = "30G"
parameter: numThreads = 1
# specify the number of causal variants
parameter: n_trait = 5
parameter: n_causal = 1
parameter: h2g = 0.05
parameter: total_h2g = False
parameter: share_pattern = "all"
parameter: independent = False
# specify the number of traits (phenotypes)
parameter: container = ""
input: genofile, group_by = 1
output: f'{cwd:a}/{step_name}/sample_{_index}_real_simulation_{n_causal}_ncausal_{n_trait}_trait.rds'
R: expand = '${ }', stdout = f"{_output:n}.stdout", stderr = f"{_output:n}.stderr", container = container
library("MASS")
library("plink2R")
library("dplyr")
library("readr")
library("tidyverse")
# source some functions to read matrix and inpute the missing data
source("~/cloud_colocalization/simulation_code/simulate_linreg.R")
source("~/cloud_colocalization/simulation_code/misc.R")
# read the plink file
simu_file = ${_input:r}
geno <- read_plink(${_input:nr})
gene_name = str_extract(simu_file, "ENSG[0-9]+")
gene_tss_map = read_tsv("/home/hs3393/coloc/fungen-xqtl-analysis/resource/gene_cis_TADB_mapper.tsv")
# filter by distance with. TSS
TSS_pos = gene_tss_map$TSS[which(gene_tss_map$gene_id == gene_name)][1]
keep_index = which(geno$bim$V4 > TSS_pos - 1500000 | geno$bim$V4 < TSS_pos + 1500000)
geno$bed <- geno$bed[, keep_index]
# Filter out columns with missing rate > 0.1
imiss <- 0.1
# Filter out columns with MAF < 0.05
maf <- 0.05
# Apply filtering
Xmat <- filter_X(geno$bed, imiss, maf)
# Only keep the first 4000 variants
ncausal <- ${n_causal}
ntrait <- ${n_trait}
indep = ${"TRUE" if independent else "FALSE"}
if (indep) {
LD_vars = 1 # Initialize LD_vars
if (ncausal == 1) {
# If only one causal variant, just sample it
vars = sample(1:ncol(Xmat), size = ncausal)
} else {
# Repeat sampling until selected variables are quasi independent
while (length(LD_vars != 0)) {
vars = sample(1:ncol(Xmat), size = ncausal)
cor_mat = cor(Xmat[, vars])
LD_vars = which(colSums(abs(cor_mat) > 0.3) > 1)
}
}
} else {
vars = sample(1:ncol(Xmat), size = ncausal)
}
# Load predefined proportions for causal variant sampling
prop <- readRDS("/home/hs3393/cloud_colocalization/simulation_code/trait6_prop.rds")[, 1:4]
proportions <- prop[ncausal, ]
# Sample variant names based on predefined proportions
sampled_name <- as.numeric(sample(names(proportions), size = ncausal, prob = proportions, replace = TRUE))
# Initialize phenotype list and effect size matrix
phenotype <- list()
B <- matrix(0, nrow = ncol(Xmat), ncol = ntrait)
# Configuration matrix for trait-variant relationships
config <- matrix(0, nrow = ntrait, ncol = ncausal)
# Assign causal variants to traits
for (i in 1:ncausal) {
coloc_trait <- sample(1:ntrait, sampled_name[i])
config[coloc_trait, i] <- 1
}
# Simulate effect sizes and phenotypes
for (i in 1:nrow(config)) {
beta <- B[, i, drop = FALSE]
index <- which(config[i, ] == 1)
if(i == 1){
h2g = ${h2g}
}else{
h2g = 0.05
}
if (length(index) > 0) {
causal_index <- vars[index]
beta <- sim_beta_fix_variant(G = Xmat, causal_index = causal_index, is_h2g_total = FALSE)
B[, i] <- beta
pheno_single <- sim_multi_traits(G = Xmat, B = as.matrix(beta), h2g = h2g, is_h2g_total = FALSE)
phenotype[[i]] <- pheno_single$P
} else {
pheno_single <- sim_multi_traits(G = Xmat, B = as.matrix(beta), h2g = h2g, is_h2g_total = FALSE)
phenotype[[i]] <- pheno_single$P
}
}
# Identify causal variants for each trait
variant <- list()
for (i in 1:ncol(B)) {
variant[[i]] <- which(B[, i] != 0)
}
# Combine phenotype data
X <- Xmat
Y <- bind_cols(phenotype)
colnames(Y) <- paste0("Trait", 1:ntrait)
# Store results in a list
data <- list()
data[["X"]] <- Xmat
data[["Y"]] <- Y
data[["variant"]] <- variant
# Save results
saveRDS(data, ${_output:r})
Simulation GWAS - 10 traits#
[GWAS_10trait]
parameter: genofile = paths
# pheno_file: give genotype file (in plink),we can read the gentype matrix. These files are separated by TADs.
parameter: cwd = path("output")
parameter: job_size = 30
parameter: walltime = "100h"
parameter: mem = "30G"
parameter: numThreads = 1
# specify the number of causal variants
parameter: n_trait = 10
parameter: n_causal = 1
parameter: h2g = 0.05
parameter: total_h2g = False
parameter: independent = False
parameter: share_pattern = "all"
# specify the number of traits (phenotypes)
parameter: container = ""
input: genofile, group_by = 1
output: f'{cwd:a}/{step_name}/sample_{_index}_real_simulation_{n_causal}_ncausal_{n_trait}_trait.rds'
R: expand = '${ }', stdout = f"{_output:n}.stdout", stderr = f"{_output:n}.stderr", container = container
library("MASS")
library("plink2R")
library("dplyr")
library("readr")
library("tidyverse")
# source some functions to read matrix and inpute the missing data
source("~/cloud_colocalization/simulation_code/simulate_linreg.R")
source("~/cloud_colocalization/simulation_code/misc.R")
# read the plink file
simu_file = ${_input:r}
geno <- read_plink(${_input:nr})
gene_name = str_extract(simu_file, "ENSG[0-9]+")
gene_tss_map = read_tsv("/home/hs3393/coloc/fungen-xqtl-analysis/resource/gene_cis_TADB_mapper.tsv")
# filter by distance with. TSS
TSS_pos = gene_tss_map$TSS[which(gene_tss_map$gene_id == gene_name)][1]
keep_index = which(geno$bim$V4 > TSS_pos - 1500000 | geno$bim$V4 < TSS_pos + 1500000)
geno$bed = geno$bed[,keep_index]
# filter out columns with missing rate > 0.1
imiss = 0.1
# filter out columns with MAF < 0.05
maf = 0.05
Xmat = filter_X(geno$bed, imiss, maf)
# only keep the first 4000 variants
ncausal = ${n_causal}
ntrait = ${n_trait}
indep = ${"TRUE" if independent else "FALSE"}
if (indep) {
LD_vars = 1 # Initialize LD_vars
if (ncausal == 1) {
# If only one causal variant, just sample it
vars = sample(1:ncol(Xmat), size = ncausal)
} else {
# Repeat sampling until selected variables are quasi independent
while (length(LD_vars != 0)) {
vars = sample(1:ncol(Xmat), size = ncausal)
cor_mat = cor(Xmat[, vars])
LD_vars = which(colSums(abs(cor_mat) > 0.3) > 1)
}
}
} else {
vars = sample(1:ncol(Xmat), size = ncausal)
}
prop = readRDS("/home/hs3393/cloud_colocalization/simulation_code/trait10_prop.rds")
proportions = prop[ncausal,]
sampled_name <- as.numeric(sample(names(proportions), size = ncausal, prob = proportions, replace = TRUE))
phenotype = list()
B = matrix(0, nrow = ncol(Xmat), ncol = ntrait)
config = matrix(0, nrow = ntrait, ncol = ncausal)
for(i in c(1:ncausal)){
coloc_trait = sample(c(1:ntrait), sampled_name[i])
config[coloc_trait, i] = 1
}
for(i in 1:nrow(config)){
beta = B[,i, drop = FALSE]
index = which(config[i,] == 1)
if(i == 1){
h2g = ${h2g}
}else{
h2g = 0.05
}
if(length(index) > 0){
causal_index = vars[index]
beta = sim_beta_fix_variant(G = Xmat, causal_index = causal_index, is_h2g_total = FALSE)
B[, i] = beta
pheno_single = sim_multi_traits(G = Xmat, B = as.matrix(beta), h2g = h2g, is_h2g_total = FALSE)
phenotype[[i]] = pheno_single$P
}else{
pheno_single = sim_multi_traits(G = Xmat, B = as.matrix(beta), h2g = h2g, is_h2g_total = FALSE)
phenotype[[i]] = pheno_single$P
}
}
variant = list()
for(i in 1:ncol(B)){
variant[[i]] = which(B[,i] != 0)
}
X = Xmat
Y = bind_cols(phenotype)
colnames(Y) = paste0("Trait", c(1:ntrait))
data = list()
data[["X"]] = Xmat
data[["Y"]] = Y
data[["variant"]] = variant
saveRDS(data, ${_output:r})
Bash submission#
work_dir="/home/hs3393/cb_Mar/simulation_data/simulation_GWAS/"
job="simulation_GWAS"
mkdir -p ${work_dir}
mkdir -p ${work_dir}/code
mkdir -p ${work_dir}/log
cd ${work_dir}/code
# Create the base_script file and write the bash code into it
cat << 'EOF' > base_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 30:00:00
#SBATCH --mem=30000
#SBATCH -J GWAS_simu
#SBATCH -o WORK_DIR/log/JOB."%j".out
#SBATCH -e WORK_DIR/log/JOB."%j".err
source ~/mamba_activate.sh
module load Singularity
sos run /home/hs3393/cb_Mar/simulation_code/6.Simulation_GWAS.ipynb JOB \
--genofile `ls /home/hs3393/cloud_colocalization/simulation_data/selected_genes_genotype/*.bim` \
--mem 30G --h2g H2G --ncausal VAR --n_trait 2 --independent \
--cwd WORK_DIR/simulation_signal_H2G/causal_VAR
EOF
base_script="base_script"
for variant in 1 2 3; do
for h2g in 0.01 0.02 0.03 0.04 0.05; do
output_script="variant_${variant}_h2g_${h2g}.sh"
cat ${base_script}|sed "s|H2G|${h2g}|g" | sed "s|VAR|${variant}|g" | sed "s|WORK_DIR|${work_dir}|g" |sed "s|JOB|${job}|g" > ${output_script}
sbatch ${output_script}
done
done
work_dir="/home/hs3393/cb_Mar/simulation_data/simulation_GWAS/GWAS_5trait/"
job="GWAS_5trait"
mkdir -p ${work_dir}
mkdir -p ${work_dir}/code
mkdir -p ${work_dir}/log
cd ${work_dir}/code
# Create the base_script file and write the bash code into it
cat << 'EOF' > base_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 30:00:00
#SBATCH --mem=30000
#SBATCH -J GWAS_simu
#SBATCH -o WORK_DIR/log/JOB."%j".out
#SBATCH -e WORK_DIR/log/JOB."%j".err
source ~/mamba_activate.sh
module load Singularity
sos run /home/hs3393/cb_Mar/simulation_code/6.Simulation_GWAS.ipynb JOB \
--genofile `ls /home/hs3393/cloud_colocalization/simulation_data/selected_genes_genotype/*.bim` \
--mem 30G --h2g H2G --ncausal VAR --n_trait 5 --independent \
--cwd WORK_DIR/simulation_signal_H2G/causal_VAR
EOF
base_script="base_script"
for variant in 1 2 3; do
for h2g in 0.02 0.03 0.04 0.05; do
output_script="variant_${variant}_h2g_${h2g}.sh"
cat ${base_script}|sed "s|H2G|${h2g}|g" | sed "s|VAR|${variant}|g" | sed "s|WORK_DIR|${work_dir}|g" |sed "s|JOB|${job}|g" > ${output_script}
sbatch ${output_script}
done
done
work_dir="/home/hs3393/cb_Mar/simulation_data/simulation_GWAS/GWAS_10trait/"
job="GWAS_10trait"
mkdir -p ${work_dir}
mkdir -p ${work_dir}/code
mkdir -p ${work_dir}/log
cd ${work_dir}/code
# Create the base_script file and write the bash code into it
cat << 'EOF' > base_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 30:00:00
#SBATCH --mem=30000
#SBATCH -J GWAS_simu
#SBATCH -o WORK_DIR/log/JOB."%j".out
#SBATCH -e WORK_DIR/log/JOB."%j".err
source ~/mamba_activate.sh
module load Singularity
sos run /home/hs3393/cb_Mar/simulation_code/6.Simulation_GWAS.ipynb JOB \
--genofile `ls /home/hs3393/cloud_colocalization/simulation_data/selected_genes_genotype/*.bim` \
--mem 30G --h2g H2G --ncausal VAR --n_trait 10 --independent \
--cwd WORK_DIR/simulation_signal_H2G/causal_VAR
EOF
base_script="base_script"
for variant in 1 2 3; do
for h2g in 0.02 0.03 0.04 0.05; do
output_script="variant_${variant}_h2g_${h2g}.sh"
cat ${base_script}|sed "s|H2G|${h2g}|g" | sed "s|VAR|${variant}|g" | sed "s|WORK_DIR|${work_dir}|g" |sed "s|JOB|${job}|g" > ${output_script}
sbatch ${output_script}
done
done
Targeted Colocboost running#
[colocboost_target]
parameter: simufile = paths
parameter: cwd = path("output")
parameter: job_size = 15
parameter: walltime = "80h"
parameter: mem = "60G"
parameter: numThreads = 3
parameter: trait = 10
parameter: container = ""
input: simufile, group_by = 1
output: f'{cwd:a}/{_input[0]:bn}_ntrait_{trait}_{step_name}.rds'
task: trunk_workers = 1, trunk_size = job_size, walltime = walltime, mem = mem, cores = numThreads, tags = f'{step_name}_{_output[0]:bn}'
R: expand = '${ }', stdout = f"{_output:n}.stdout", stderr = f"{_output:n}.stderr", container = container
for(file in list.files("/home/xc2270/COLOCBoost/code_COLOCBoost/colocboost_updating/", full.names = T)) {source(file)}
X = list()
Y = list()
variant = list()
rds = readRDS(${_input:ar})
for(i in 1:${trait}){
X[[i]] = rds$X
Y[[i]] = as.matrix(rds$Y[, i, drop = FALSE])
variant[[i]] = rds$variant[[i]]
}
start_time <- Sys.time()
colocboost_result = colocboost(
X = X,
Y = Y, target_outcome_idx=1
)
end_time <- Sys.time()
# record true variant, analysed trait number and corresponding file name
colocboost_result$var = variant
colocboost_result$trait_num = ${trait}
colocboost_result$file = "${_input[0]:a}"
# In real setting (list: variant), show which snp appear in at least two traits
all_var = unlist(variant)
true_var = as.numeric(names(which(table(all_var) >= 2)))
true_trait = list()
# Iterate through the variant list to find which traits the true_var is colocalized on
for (variant_index in 1:length(true_var)){
temp_vec = c()
for(i in 1:length(variant)){
if(true_var[variant_index] %in% variant[[i]]){
temp_vec = c(temp_vec, i)
}
}
true_trait[[variant_index]] = temp_vec
}
library(stringr)
coloc_trait = list()
# if no coloc sets detected, assign coloc_trait as NULL as well
if(length(colocboost_result$cos_details$cos$cos_index) == 0){
coloc_trait = NULL
} else {
for(i in 1:length(colocboost_result$cos_details$cos_outcomes$outcome_index)){
coloc_trait[[i]] = colocboost_result$cos_details$cos_outcomes$outcome_index[[i]]
}
}
colocboost_result$true_variant = true_var
colocboost_result$true_trait = true_trait
colocboost_result$coloc_set = colocboost_result$cos_details$cos$cos_index
colocboost_result$coloc_trait = coloc_trait
colocboost_result$time = end_time - start_time
saveRDS(colocboost_result, ${_output:r})
Bash job submission#
data_dir="/home/hs3393/cb_Mar/simulation_data/simulation_GWAS/"
job="colocboost_target"
work_dir="/home/hs3393/cb_Mar/simulation_result/simulation_GWAS/"
#!/bin/bash
mkdir -p ${work_dir}/${job}/code
mkdir -p ${work_dir}/${job}/log
mkdir -p ${work_dir}/${job}/result
cd ${work_dir}/${job}/code
cat << 'EOF' > base_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 100:00:00
#SBATCH --mem=30000
#SBATCH -J JOB
#SBATCH -o WORK_DIR/JOB/log/JOB.%j.out
#SBATCH -e WORK_DIR/JOB/log/JOB.%j.err
source /home/hs3393/mamba_activate.sh
module load Singularity
cd DATA_DIR/simulation_signal_H2G/causal_VAR
sos run /home/hs3393/cb_Mar/simulation_code/6.Simulation_GWAS.ipynb JOB \
--simufile $(find -type f -name '*.rds') \
--mem 30G --trait 2 \
--cwd WORK_DIR/JOB/simulation_signal_H2G/causal_VAR
EOF
base_script="base_script"
for variant in 1 2 3; do
for h2g in 0.01 0.02 0.03 0.04 0.05; do
output_script="variant_${variant}_h2g_${h2g}.sh"
cat ${base_script}|sed "s|H2G|${h2g}|g" | sed "s|VAR|${variant}|g" | sed "s|WORK_DIR|${work_dir}|g" | sed "s|DATA_DIR|${data_dir}|g" |sed "s|JOB|${job}|g" > ${output_script}
sbatch ${output_script}
done
done
data_dir="/home/hs3393/cb_Mar/simulation_data/simulation_GWAS/"
job="colocboost"
work_dir="/home/hs3393/cb_Mar/simulation_result/simulation_GWAS/"
#!/bin/bash
mkdir -p ${work_dir}/${job}/code
mkdir -p ${work_dir}/${job}/log
mkdir -p ${work_dir}/${job}/result
cd ${work_dir}/${job}/code
cat << 'EOF' > base_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 100:00:00
#SBATCH --mem=30000
#SBATCH -J JOB
#SBATCH -o WORK_DIR/JOB/log/JOB.%j.out
#SBATCH -e WORK_DIR/JOB/log/JOB.%j.err
source /home/hs3393/mamba_activate.sh
module load Singularity
cd DATA_DIR/simulation_signal_H2G/causal_VAR
sos run /home/hs3393/cb_Mar/simulation_code/2.Run_Colocboost.ipynb JOB \
--simufile $(find -type f -name '*.rds') \
--mem 30G --trait 2 \
--cwd WORK_DIR/JOB/simulation_signal_H2G/causal_VAR
EOF
base_script="base_script"
for variant in 1 2 3; do
for h2g in 0.02 0.03 0.04 0.05; do
output_script="variant_${variant}_h2g_${h2g}.sh"
cat ${base_script}|sed "s|H2G|${h2g}|g" | sed "s|VAR|${variant}|g" | sed "s|WORK_DIR|${work_dir}|g" | sed "s|DATA_DIR|${data_dir}|g" |sed "s|JOB|${job}|g" > ${output_script}
sbatch ${output_script}
done
done
Result summary#
data_dir="/home/hs3393/cb_Mar/simulation_result/simulation_GWAS/colocboost_target/"
mkdir -p ${data_dir}/summary
cd ${data_dir}/summary
cat << 'EOF' > summary_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 8:00:00
#SBATCH --mem=30000
#SBATCH -J sum
#SBATCH -o DATA_DIR/log/summary."%j".out
#SBATCH -e DATA_DIR/log/summary."%j".err
source ~/mamba_activate.sh
sos run /home/hs3393/cb_Mar/simulation_code/4.Result_Summary.ipynb coloc_summary \
--folder DATA_DIR/ \
--cwd DATA_DIR/summary
EOF
base_script="summary_script"
output_script="summary.sh"
cat ${base_script}| sed "s|DATA_DIR|${data_dir}|g" > ${output_script}
sbatch ${output_script}
data_dir="/home/hs3393/cb_Mar/simulation_result/simulation_GWAS/colocboost/"
mkdir -p ${data_dir}/summary
cd ${data_dir}/summary
cat << 'EOF' > summary_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 8:00:00
#SBATCH --mem=30000
#SBATCH -J sum
#SBATCH -o DATA_DIR/log/summary."%j".out
#SBATCH -e DATA_DIR/log/summary."%j".err
source ~/mamba_activate.sh
sos run /home/hs3393/cb_Mar/simulation_code/4.Result_Summary.ipynb coloc_summary \
--folder DATA_DIR/ \
--cwd DATA_DIR/summary
EOF
base_script="summary_script"
output_script="summary.sh"
cat ${base_script}| sed "s|DATA_DIR|${data_dir}|g" > ${output_script}
sbatch ${output_script}
data_dir="/home/hs3393/cb_simulation/simulation_result/colocboost/simulation_GWAS_not_targeted/"
mkdir -p ${data_dir}/summary
cd ${data_dir}/summary
cat << 'EOF' > summary_script
#!/bin/bash -l
# NOTE the -l flag!
#
#SBATCH -t 8:00:00
#SBATCH --mem=30000
#SBATCH -J sum
#SBATCH -o DATA_DIR/log/summary."%j".out
#SBATCH -e DATA_DIR/log/summary."%j".err
source ~/mamba_activate.sh
sos run /home/hs3393/cb_Mar/simulation_code/4.Result_Summary.ipynb coloc_summary \
--folder DATA_DIR/ \
--cwd DATA_DIR/summary
EOF
base_script="summary_script"
output_script="summary.sh"
cat ${base_script}| sed "s|DATA_DIR|${data_dir}|g" > ${output_script}
sbatch ${output_script}