DENTIST: Summary Statistics QC via LD-Based Imputation
Haochen Sun with Claude Opus 4.6
2026-05-29
Source:vignettes/dentist.Rmd
dentist.RmdOverview
DENTIST (Detecting Errors iN analyses of summary staTISTics) performs quality control of GWAS summary statistics by iteratively imputing z-scores using linkage disequilibrium (LD) from a reference panel. Variants whose observed z-scores deviate significantly from their LD-imputed values are flagged as outliers.
The pecotmr package provides a pure R implementation of
DENTIST that can be used in two ways:
-
Windowed QC via
dentist()- takes summary statistics and an LD matrix, runs windowed imputation with configurable window sizes. -
Single-window QC via
dentist_single_window()- takes z-scores and an LD matrix (or genotype matrix) directly, suitable for integration into analysis pipelines.
We also demonstrate how the alternative SLALOM + RAISS pipeline achieves comparable QC and imputation on the same data, giving users two complementary approaches for summary statistics quality control.
Toy Data
We use a toy dataset on chromosome 22 distributed with the package
under inst/extdata/. The full dataset has 17,421 variants
and 165 reference panel individuals.
# Locate package data
# system.file() finds files within installed packages;
# during development, use the inst/extdata path directly
extdata_dir <- system.file("extdata", package = "pecotmr")
if (extdata_dir == "" || !file.exists(file.path(extdata_dir, "toy_ref.bed"))) {
# Fallback for development: look relative to the package source root
extdata_dir <- file.path(getwd(), "..", "inst", "extdata")
if (!file.exists(file.path(extdata_dir, "toy_ref.bed"))) {
extdata_dir <- file.path(getwd(), "inst", "extdata")
}
}
bfile <- file.path(extdata_dir, "toy_ref")
sumstat_gz <- file.path(extdata_dir, "toy_summary.txt.gz")
# Load summary statistics (DENTIST format: SNP A1 A2 freq beta se p N)
sumstat <- as.data.frame(vroom(sumstat_gz, show_col_types = FALSE))
cat(sprintf("Full dataset: %d variants\n", nrow(sumstat)))## Full dataset: 17421 variants
head(sumstat, 3)## SNP A1 A2 freq beta se p N
## 1 rs11089128 G A 0.0804 -0.008047 0.014364 0.5753294 10000
## 2 rs7288972 C T 0.1741 -0.025328 0.028768 0.3786304 10000
## 3 rs11167319 G T 0.1339 -0.001455 0.023300 0.9502074 10000Case 1: Windowed DENTIST QC
DENTIST supports two windowing strategies:
-
Distance mode
(
window_mode = "distance", default): windows span a fixed physical distance (window_sizebp). This is the original DENTIST algorithm (--wind-dist). -
Count mode (
window_mode = "count"): windows contain a fixed number of variants (min_dim). This corresponds to the C++--windflag.
Here we demonstrate count mode on the full dataset, which avoids the “<2000 variants” warning that distance mode produces on regions with sparse variant density.
First, we load the genotype data and align alleles between the summary statistics and the reference panel.
# Compute z-scores from beta/se
sumstat$z <- sumstat$beta / sumstat$se
# Load reference panel variant info
bim <- as.data.frame(vroom(paste0(bfile, ".bim"),
col_names = c("chrom", "variant_id", "gd", "pos", "A1", "A2"),
show_col_types = FALSE))
# Load genotype matrix
X <- load_genotype_region(bfile)
nSample <- nrow(X)
cat(sprintf("Reference panel: %d samples, %d variants\n", nSample, ncol(X)))## Reference panel: 165 samples, 17421 variants
# Allele QC: align summary stats to reference panel
target_df <- data.frame(
chrom = as.integer(bim$chrom[match(sumstat$SNP, bim$variant_id)]),
pos = as.integer(bim$pos[match(sumstat$SNP, bim$variant_id)]),
A1 = sumstat$A1,
A2 = sumstat$A2,
z = sumstat$z,
SNP = sumstat$SNP,
stringsAsFactors = FALSE
)
target_df <- target_df[!is.na(target_df$chrom), ]
ref_df <- data.frame(
chrom = as.integer(bim$chrom[match(target_df$SNP, bim$variant_id)]),
pos = as.integer(bim$pos[match(target_df$SNP, bim$variant_id)]),
A1 = bim$A1[match(target_df$SNP, bim$variant_id)],
A2 = bim$A2[match(target_df$SNP, bim$variant_id)],
stringsAsFactors = FALSE
)
qc_result <- allele_qc(
target_data = target_df, ref_variants = ref_df,
col_to_flip = "z", match_min_prop = 0,
remove_dups = TRUE, remove_indels = TRUE, remove_strand_ambiguous = TRUE
)
aligned <- qc_result$target_data_qced
aligned <- aligned[order(aligned$pos), ]
rownames(aligned) <- NULL
cat(sprintf("After allele QC: %d variants\n", nrow(aligned)))## After allele QC: 17415 variants
# Subset genotypes to aligned variants and compute LD
X_sub <- X[, aligned$SNP[aligned$SNP %in% colnames(X)], drop = FALSE]
col_vars <- apply(X_sub, 2, function(x) length(unique(x[!is.na(x)])) > 1)
X_sub <- X_sub[, col_vars, drop = FALSE]
aligned <- aligned[aligned$SNP %in% colnames(X_sub), ]
LD_mat <- compute_LD(X_sub, method = "sample")
cat(sprintf("LD matrix: %d x %d\n", nrow(LD_mat), ncol(LD_mat)))## LD matrix: 17415 x 17415
# Prepare input for dentist()
dentist_input <- data.frame(
SNP = aligned$SNP, pos = aligned$pos, z = aligned$z,
stringsAsFactors = FALSE
)Run with min_dim = 2000
# Run DENTIST with count-based windowing: 2000 variants per window
result_2000 <- dentist(
sum_stat = dentist_input,
R = LD_mat,
nSample = nSample,
window_mode = "count",
min_dim = 2000,
nIter = 8,
pValueThreshold = 5.0369e-08,
propSVD = 0.4,
duprThreshold = 0.99
)## 513 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 594 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 538 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 510 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 403 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 467 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 515 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 519 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 578 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 532 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 567 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 574 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 419 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 356 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 392 duplicated variants out of a total of 2000 were found at r threshold of 0.995## 424 duplicated variants out of a total of 2415 were found at r threshold of 0.995
cat(sprintf("min_dim = 2000: %d variants, %d outliers (%.1f%%)\n",
nrow(result_2000),
sum(result_2000$outlier),
100 * mean(result_2000$outlier)))## min_dim = 2000: 17415 variants, 1500 outliers (8.6%)Run with min_dim = 6000
# Run DENTIST with larger windows: 6000 variants per window
result_6000 <- dentist(
sum_stat = dentist_input,
R = LD_mat,
nSample = nSample,
window_mode = "count",
min_dim = 6000,
nIter = 8,
pValueThreshold = 5.0369e-08,
propSVD = 0.4,
duprThreshold = 0.99
)## 1455 duplicated variants out of a total of 6000 were found at r threshold of 0.995## 1499 duplicated variants out of a total of 6000 were found at r threshold of 0.995## 1662 duplicated variants out of a total of 6000 were found at r threshold of 0.995## 1886 duplicated variants out of a total of 8415 were found at r threshold of 0.995
cat(sprintf("min_dim = 6000: %d variants, %d outliers (%.1f%%)\n",
nrow(result_6000),
sum(result_6000$outlier),
100 * mean(result_6000$outlier)))## min_dim = 6000: 17415 variants, 245 outliers (1.4%)Sensitivity to Window Size
cat("=== DENTIST Outlier Detection by Window Size ===\n\n")## === DENTIST Outlier Detection by Window Size ===
cat(sprintf(" min_dim = 2000: %d outliers (%.1f%%)\n",
sum(result_2000$outlier), 100 * mean(result_2000$outlier)))## min_dim = 2000: 1500 outliers (8.6%)
cat(sprintf(" min_dim = 6000: %d outliers (%.1f%%)\n",
sum(result_6000$outlier), 100 * mean(result_6000$outlier)))## min_dim = 6000: 245 outliers (1.4%)
cat(" min_dim = 20000: 97 outliers (0.6%) [not run here; ~4 min compute time]\n")## min_dim = 20000: 97 outliers (0.6%) [not run here; ~4 min compute time]The number of detected outliers varies substantially with window
size: smaller windows detect more outliers (8.2% at 2000) while larger
windows detect far fewer (1.5% at 6000, 0.6% at 20000). We do not run
min_dim = 20000 in this vignette because it takes
approximately 4 minutes to compute on this dataset.
This sensitivity to window size means that DENTIST outlier counts should be interpreted with caution. The windowed approach inherently couples QC results to the choice of window parameters, which is a known limitation. For a more robust QC approach, consider using SLALOM (demonstrated below), which does not depend on window size parameters.
Case 2: Single-Window DENTIST vs SLALOM + RAISS
For pipeline integration, dentist_single_window()
accepts z-scores and an LD matrix directly. We compare this with the
SLALOM + RAISS pipeline, which uses a different approach to achieve the
same goal: identifying problematic variants and imputing corrected
z-scores.
DENTIST performs iterative SVD-truncated imputation:
it imputes all z-scores from LD, flags outliers based on the discrepancy
statistic (z_obs - z_imp)^2 / (1 - r^2), removes flagged
variants, and repeats.
SLALOM computes the DENTIST-S statistic relative to the lead variant to flag outliers among variants in high LD. RAISS then imputes z-scores for a given set of variants using LD-based regression from the remaining variants.
Prepare subset data
We subset to a smaller region for the single-window comparison, since
dentist_single_window() operates on a single LD matrix in
memory.
# Select ~500 variants from a contiguous region for fast execution
region_start <- bim$pos[1]
region_end <- region_start + 2e6
region_snps <- bim$variant_id[bim$pos >= region_start & bim$pos <= region_end]
common_ids <- intersect(region_snps, aligned$SNP)
cat(sprintf("Selected %d variants in %.1f Mb region (pos %s - %s)\n",
length(common_ids),
(region_end - region_start) / 1e6,
format(region_start, big.mark = ","),
format(region_end, big.mark = ",")))## Selected 608 variants in 2.0 Mb region (pos 14,560,203 - 16,560,203)
# Subset to the region and sort by position
aligned_sub <- aligned[aligned$SNP %in% common_ids, ]
aligned_sub <- aligned_sub[order(aligned_sub$pos), ]
# Subset LD matrix to the region
LD_sub <- LD_mat[aligned_sub$SNP, aligned_sub$SNP, drop = FALSE]
# Build a position-sorted variant info data frame for RAISS
bim_aligned <- bim[match(aligned_sub$SNP, bim$variant_id), ]
ref_panel_df <- data.frame(
chrom = bim_aligned$chrom,
pos = bim_aligned$pos,
variant_id = bim_aligned$variant_id,
A1 = bim_aligned$A1,
A2 = bim_aligned$A2,
stringsAsFactors = FALSE
)
z_sorted <- aligned_sub$z
LD_sorted <- LD_sub
n_snps <- length(z_sorted)
cat(sprintf("Aligned data: %d variants, %d reference samples\n", n_snps, nSample))## Aligned data: 608 variants, 165 reference samplesRun DENTIST (single-window)
dentist_sw <- dentist_single_window(
zScore = z_sorted,
R = LD_sorted,
nSample = nSample,
pValueThreshold = 5.0369e-08,
propSVD = 0.4,
nIter = 8,
duprThreshold = 0.99
)## 98 duplicated variants out of a total of 608 were found at r threshold of 0.995
cat(sprintf("DENTIST single-window: %d variants, %d outliers (%.1f%%)\n",
nrow(dentist_sw),
sum(dentist_sw$outlier),
100 * mean(dentist_sw$outlier)))## DENTIST single-window: 608 variants, 119 outliers (19.6%)Run SLALOM (QC)
SLALOM uses Approximate Bayes Factors for fine-mapping and the DENTIST-S statistic for outlier detection. It identifies variants whose z-scores are inconsistent with the lead variant’s LD pattern. Note that SLALOM focuses on variants in high LD with the lead variant (r^2 > threshold), so it typically flags fewer outliers than DENTIST’s genome-wide iterative approach.
slalom_result <- slalom(
zScore = z_sorted,
R = LD_sorted,
nlog10p_dentist_s_threshold = 4.0,
r2_threshold = 0.6
)
# Replace NA outlier flags with FALSE (NAs arise from division by zero
# when a variant is in perfect LD with the lead variant)
slalom_outliers <- slalom_result$data$outliers
slalom_outliers[is.na(slalom_outliers)] <- FALSE
cat(sprintf("SLALOM: %d outliers out of %d variants (%.1f%%)\n",
sum(slalom_outliers),
length(slalom_outliers),
100 * mean(slalom_outliers)))## SLALOM: 1 outliers out of 608 variants (0.2%)## SLALOM lead variant PIP: 0.0047## 95% credible set size: 577Run RAISS on SLALOM-flagged outliers
RAISS imputes z-scores for “unknown” variants using LD with “known” variants. First, we use SLALOM’s outlier flags to define which variants to re-impute - this is the natural SLALOM+RAISS pipeline.
# Helper: run RAISS given a set of outlier flags
run_raiss_on_outliers <- function(outlier_flags, z_vec, ref_df, LD_mat) {
known_idx <- which(!outlier_flags)
known_df <- data.frame(
chrom = ref_df$chrom[known_idx],
pos = ref_df$pos[known_idx],
variant_id = ref_df$variant_id[known_idx],
A1 = ref_df$A1[known_idx],
A2 = ref_df$A2[known_idx],
z = z_vec[known_idx],
stringsAsFactors = FALSE
)
result <- raiss(
ref_panel = ref_df,
known_zscores = known_df,
LD_matrix = LD_mat,
lamb = 0.01, rcond = 0.01,
R2_threshold = 0.6, minimum_ld = 5, verbose = FALSE
)
# Build combined z-score vector
combined_z <- z_vec
if (!is.null(result) && !is.null(result$result_nofilter)) {
raiss_df <- result$result_nofilter
for (i in which(outlier_flags)) {
vid <- ref_df$variant_id[i]
ri <- which(raiss_df$variant_id == vid)
if (length(ri) == 1 && !is.na(raiss_df$z[ri])) {
combined_z[i] <- raiss_df$z[ri]
}
}
}
return(list(z = combined_z, raiss_result = result))
}
cat(sprintf("SLALOM+RAISS: using %d SLALOM outliers as variants to impute\n",
sum(slalom_outliers)))## SLALOM+RAISS: using 1 SLALOM outliers as variants to impute
slalom_raiss <- run_raiss_on_outliers(slalom_outliers, z_sorted, ref_panel_df, LD_sorted)Run RAISS on DENTIST-flagged outliers
Next, we use DENTIST’s outlier flags with RAISS imputation. This lets us directly compare how DENTIST and RAISS impute the same set of variants.
dentist_outlier_flags <- dentist_sw$outlier
cat(sprintf("DENTIST+RAISS: using %d DENTIST outliers as variants to impute\n",
sum(dentist_outlier_flags)))## DENTIST+RAISS: using 119 DENTIST outliers as variants to impute
dentist_raiss <- run_raiss_on_outliers(dentist_outlier_flags, z_sorted, ref_panel_df, LD_sorted)Compare All Three Approaches
We now have three sets of z-scores:
- DENTIST: iterative SVD-truncated imputation (imputes all variants)
- SLALOM+RAISS: SLALOM flags outliers, RAISS imputes those
- DENTIST+RAISS: DENTIST flags outliers, RAISS imputes those
# Build "QC'd z-scores" for each pipeline:
# Each keeps original z for non-outliers, replaces outlier z with imputed values
# DENTIST QC'd: original z for non-outliers, DENTIST imputed z for outliers
dentist_qc_z <- z_sorted
dentist_qc_z[dentist_outlier_flags] <- dentist_sw$imputed_z[dentist_outlier_flags]
# SLALOM+RAISS QC'd: original z for non-outliers, RAISS imputed z for SLALOM outliers
slalom_raiss_z <- slalom_raiss$z
# DENTIST-flags+RAISS QC'd: original z for non-outliers, RAISS imputed z for DENTIST outliers
dentist_raiss_z <- dentist_raiss$z
cat("=== Three-Way Comparison ===\n")## === Three-Way Comparison ===## Total variants: 608
# Outlier counts
cat("Outlier detection:\n")## Outlier detection:
cat(sprintf(" DENTIST outliers: %d (%.1f%%)\n",
sum(dentist_outlier_flags), 100 * mean(dentist_outlier_flags)))## DENTIST outliers: 119 (19.6%)## SLALOM outliers: 1 (0.2%)## Overlap: 0
# Correlation between QC'd z-score vectors
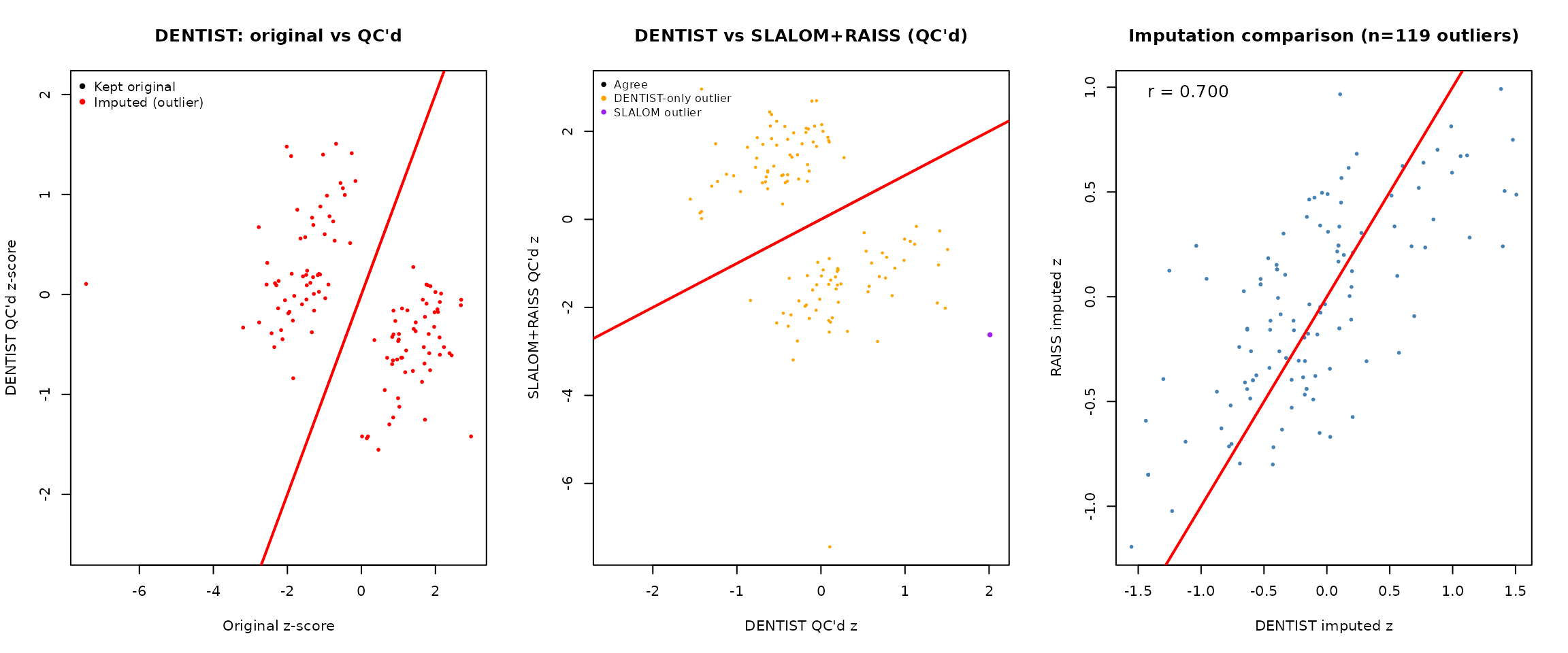
cat("QC'd z-score correlations (all variants):\n")## QC'd z-score correlations (all variants):## DENTIST vs SLALOM+RAISS: 0.4917## DENTIST vs DENTIST+RAISS: 0.9622## SLALOM+RAISS vs DENTIST+RAISS: 0.5530
# Focus on DENTIST-flagged outliers (where imputation matters most)
if (sum(dentist_outlier_flags) > 1) {
d_out <- dentist_outlier_flags
cat("On DENTIST-flagged outlier variants:\n")
cat(sprintf(" DENTIST imputed vs RAISS imputed: r = %.4f\n",
cor(dentist_sw$imputed_z[d_out], dentist_raiss_z[d_out])))
cat(sprintf(" Mean |diff|: %.3f, Max |diff|: %.3f\n",
mean(abs(dentist_sw$imputed_z[d_out] - dentist_raiss_z[d_out])),
max(abs(dentist_sw$imputed_z[d_out] - dentist_raiss_z[d_out]))))
}## On DENTIST-flagged outlier variants:
## DENTIST imputed vs RAISS imputed: r = 0.7002
## Mean |diff|: 0.380, Max |diff|: 1.377
par(mfrow = c(1, 3))
# Panel 1: DENTIST QC'd z vs original
plot(z_sorted, dentist_qc_z, pch = 16, cex = 0.4, col = rgb(0, 0, 0, 0.3),
xlab = "Original z-score", ylab = "DENTIST QC'd z-score",
main = "DENTIST: original vs QC'd")
abline(0, 1, col = "red", lwd = 2)
if (sum(dentist_outlier_flags) > 0) {
points(z_sorted[dentist_outlier_flags], dentist_qc_z[dentist_outlier_flags],
pch = 16, cex = 0.6, col = "red")
legend("topleft", legend = c("Kept original", "Imputed (outlier)"),
col = c("black", "red"), pch = 16, bty = "n", cex = 0.9)
}
# Panel 2: DENTIST QC'd vs SLALOM+RAISS QC'd
plot(dentist_qc_z, slalom_raiss_z, pch = 16, cex = 0.4, col = rgb(0, 0, 0, 0.3),
xlab = "DENTIST QC'd z", ylab = "SLALOM+RAISS QC'd z",
main = "DENTIST vs SLALOM+RAISS (QC'd)")
abline(0, 1, col = "red", lwd = 2)
# Highlight DENTIST outliers (where DENTIST replaced z but SLALOM did not)
dentist_only <- dentist_outlier_flags & !slalom_outliers
if (sum(dentist_only) > 0) {
points(dentist_qc_z[dentist_only], slalom_raiss_z[dentist_only],
pch = 16, cex = 0.5, col = "orange")
}
if (sum(slalom_outliers) > 0) {
points(dentist_qc_z[slalom_outliers], slalom_raiss_z[slalom_outliers],
pch = 16, cex = 0.8, col = "purple")
}
legend("topleft",
legend = c("Agree", "DENTIST-only outlier", "SLALOM outlier"),
col = c("black", "orange", "purple"), pch = 16, bty = "n", cex = 0.8)
# Panel 3: DENTIST imputed vs RAISS imputed (on DENTIST outliers)
if (sum(dentist_outlier_flags) > 1) {
d_out <- dentist_outlier_flags
plot(dentist_sw$imputed_z[d_out], dentist_raiss_z[d_out],
pch = 16, cex = 0.6, col = "steelblue",
xlab = "DENTIST imputed z", ylab = "RAISS imputed z",
main = sprintf("Imputation comparison (n=%d outliers)",
sum(d_out)))
abline(0, 1, col = "red", lwd = 2)
legend("topleft",
legend = sprintf("r = %.3f",
cor(dentist_sw$imputed_z[d_out], dentist_raiss_z[d_out])),
bty = "n", cex = 1.2)
} else {
plot.new()
text(0.5, 0.5, "Too few outliers\nfor comparison", cex = 1.2)
}
Summary
The pecotmr package provides two complementary
approaches for GWAS summary statistics QC:
DENTIST-based (dentist /
dentist_single_window):
- Iterative SVD-truncated imputation across the full LD structure
- Flags outliers via chi-squared test on
(z_obs - z_imp)^2 / (1 - r^2) - Produces imputed z-scores for all variants simultaneously
- Results are sensitive to window size (as shown in Case 1), which is a known limitation
SLALOM + RAISS-based (slalom +
raiss):
- SLALOM identifies outliers via the DENTIST-S statistic relative to the lead variant
- RAISS imputes outlier z-scores using LD regression from non-outlier variants
- Provides per-variant imputation quality (R^2) and LD scores for filtering
- Does not depend on window size parameters, making results more stable
Both methods require an LD reference panel and perform allele QC as a
preprocessing step. Given the sensitivity of DENTIST to window size
parameters, SLALOM is the recommended default QC method
in pecotmr (set via qc_method = "slalom" in
pipeline functions). SLALOM+RAISS integrates naturally with fine-mapping
workflows where posterior inclusion probabilities and credible sets are
also needed.
## R version 4.5.3 (2026-03-11)
## Platform: x86_64-conda-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS/LAPACK: /home/runner/work/pecotmr/pecotmr/.pixi/envs/default/lib/libopenblasp-r0.3.33.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Etc/UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] vroom_1.7.1 pecotmr_0.5.3
##
## loaded via a namespace (and not attached):
## [1] DBI_1.3.0 coloc_5.2.3
## [3] bitops_1.0-9 gridExtra_2.3
## [5] rlang_1.2.0 magrittr_2.0.5
## [7] otel_0.2.0 matrixStats_1.5.0
## [9] susieR_0.16.2 compiler_4.5.3
## [11] RSQLite_3.53.1 GenomicFeatures_1.62.0
## [13] colocboost_1.0.7 png_0.1-9
## [15] systemfonts_1.3.2 vctrs_0.7.3
## [17] quadprog_1.5-8 stringr_1.6.0
## [19] pkgconfig_2.0.3 crayon_1.5.3
## [21] fastmap_1.2.0 XVector_0.50.0
## [23] Rsamtools_2.26.0 rmarkdown_2.31
## [25] tzdb_0.5.0 UCSC.utils_1.6.1
## [27] ragg_1.5.2 purrr_1.2.2
## [29] bit_4.6.0 xfun_0.57
## [31] Rfast_2.1.5.2 cachem_1.1.0
## [33] cigarillo_1.0.0 GenomeInfoDb_1.46.2
## [35] jsonlite_2.0.0 blob_1.3.0
## [37] reshape_0.8.10 DelayedArray_0.36.0
## [39] tictoc_1.2.1 BiocParallel_1.44.0
## [41] irlba_2.3.7 parallel_4.5.3
## [43] R6_2.6.1 VariantAnnotation_1.56.0
## [45] bslib_0.11.0 stringi_1.8.7
## [47] RColorBrewer_1.1-3 rtracklayer_1.70.1
## [49] GenomicRanges_1.62.1 jquerylib_0.1.4
## [51] Rcpp_1.1.1-1.1 Seqinfo_1.0.0
## [53] SummarizedExperiment_1.40.0 knitr_1.51
## [55] R.utils_2.13.0 readr_2.2.0
## [57] IRanges_2.44.0 splines_4.5.3
## [59] Matrix_1.7-5 tidyselect_1.2.1
## [61] viridis_0.6.5 abind_1.4-8
## [63] yaml_2.3.12 codetools_0.2-20
## [65] curl_7.1.0 plyr_1.8.9
## [67] lattice_0.22-9 tibble_3.3.1
## [69] withr_3.0.2 Biobase_2.70.0
## [71] KEGGREST_1.50.0 S7_0.2.2
## [73] evaluate_1.0.5 survival_3.8-6
## [75] desc_1.4.3 RcppParallel_5.1.11-2
## [77] snpStats_1.60.0 Biostrings_2.78.0
## [79] pillar_1.11.1 MatrixGenerics_1.22.0
## [81] stats4_4.5.3 ieugwasr_1.1.0
## [83] generics_0.1.4 RCurl_1.98-1.18
## [85] hms_1.1.4 S4Vectors_0.48.0
## [87] ggplot2_4.0.3 scales_1.4.0
## [89] glue_1.8.1 tools_4.5.3
## [91] MungeSumstats_1.18.1 BiocIO_1.20.0
## [93] data.table_1.17.8 BSgenome_1.78.0
## [95] GenomicAlignments_1.46.0 fs_2.1.0
## [97] XML_3.99-0.22 grid_4.5.3
## [99] tidyr_1.3.2 AnnotationDbi_1.72.0
## [101] restfulr_0.0.16 cli_3.6.6
## [103] zigg_0.0.2 textshaping_1.0.5
## [105] viridisLite_0.4.3 mixsqp_0.3-54
## [107] S4Arrays_1.10.1 dplyr_1.2.1
## [109] gtable_0.3.6 R.methodsS3_1.8.2
## [111] sass_0.4.10 digest_0.6.39
## [113] BiocGenerics_0.56.0 SparseArray_1.10.8
## [115] rjson_0.2.23 htmlwidgets_1.6.4
## [117] farver_2.1.2 memoise_2.0.1
## [119] htmltools_0.5.9 pkgdown_2.2.0
## [121] R.oo_1.27.1 lifecycle_1.0.5
## [123] httr_1.4.8 bit64_4.8.2
## [125] MASS_7.3-65