Adjusting for covariates in functional fine-mapping
William Denault
Source:vignettes/fsusie_covariates_adjustment.Rmd
fsusie_covariates_adjustment.RmdThis vignette adjusts a functional response Y for
covariate effects before fine-mapping. Observed covariates that affect
the trait at every position (age, sex, batch, principal components of a
global expression matrix) inflate the residual variance and attenuate
the genetic signal if left in. The right tool for removing such effects
on a functional response is a wavelet- domain empirical-Bayes regression
that accommodates smooth position-dependent covariate effects.
mfsusieR exposes this through
mf_adjust_for_covariates().
Generating the data
We simulate functional responses with two ingredients:
-
Genetic signal — two causal SNPs (positions 25 and
75 in the genotype matrix) with smooth random per-position effects
f1,f2. -
Covariate signal — three covariates (independent of
genotype) acting through smooth random per-position coefficient curves
f1_cov,f2_cov,f3_cov.
All effect curves are drawn from
mf_simu_ibss_per_level(7)$sim_func, which samples from the
IBSS wavelet prior (the same prior class fSuSiE infers).
rsnr <- 1
pos1 <- 25L
pos2 <- 75L
lev_res <- 7L
T_m <- 2L^lev_res
f1 <- mf_simu_ibss_per_level(lev_res)$sim_func
f2 <- mf_simu_ibss_per_level(lev_res)$sim_func
f1_cov <- mf_simu_ibss_per_level(lev_res)$sim_func
f2_cov <- mf_simu_ibss_per_level(lev_res)$sim_func
f3_cov <- mf_simu_ibss_per_level(lev_res)$sim_func
plot(f1, type = "l", col = "royalblue", lwd = 2,
xlab = "position", ylab = "effect")
abline(h = 0, lty = 3, col = "grey50")
lines(f2, type = "l", col = "limegreen", lwd = 2)
legend("topright", legend = c("f1 (SNP 25)", "f2 (SNP 75)"),
col = c("royalblue", "limegreen"), lwd = 2, bty = "n")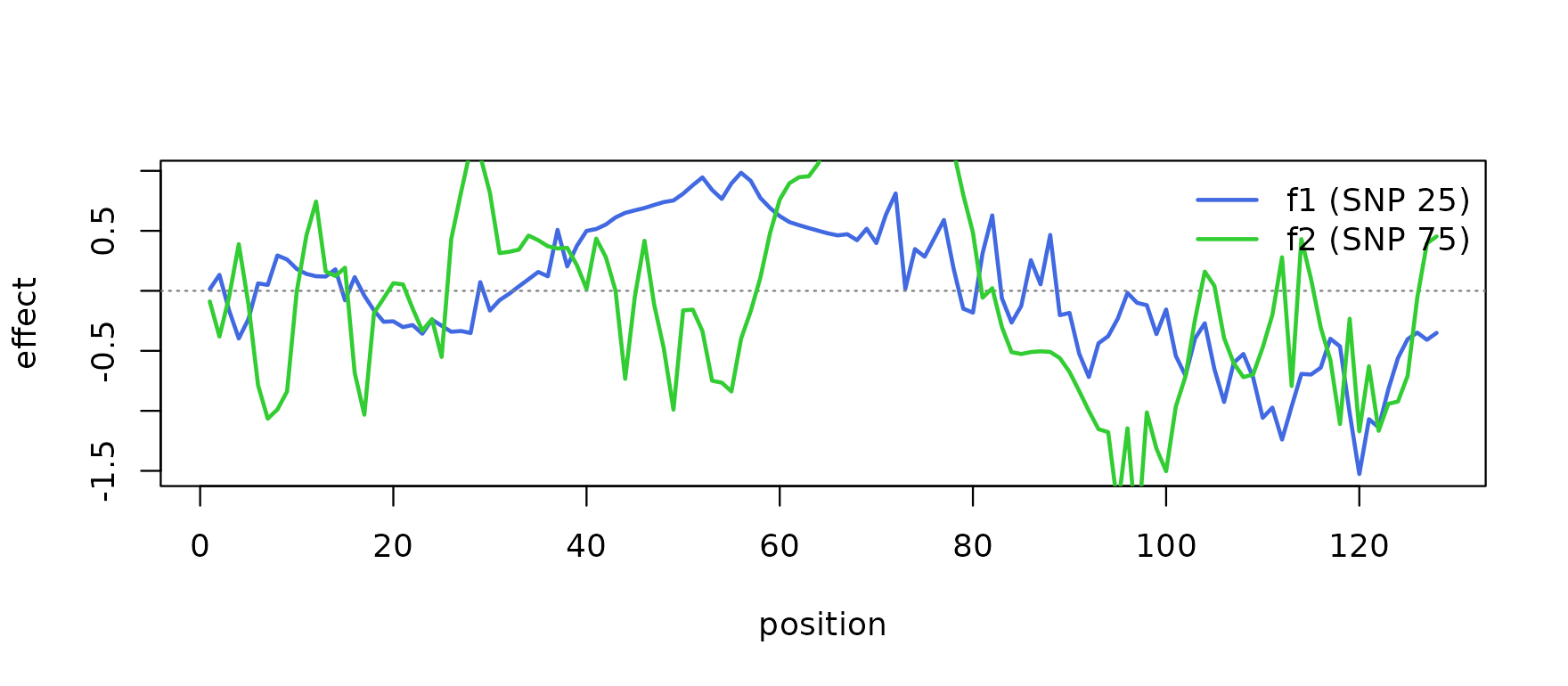
The observed data is a mixture of covariate-driven confounding and
genotype signal. genetic_signal is the part we want to
recover; covariate_signal is the part we want to
remove.
data(N3finemapping)
Geno <- N3finemapping$X[, seq_len(100)]
n <- nrow(Geno)
# Three covariates, sd = 2; independent of Geno.
Cov <- matrix(rnorm(3L * n, sd = 2), ncol = 3L)
genetic_signal <- matrix(0, n, T_m)
covariate_signal <- matrix(0, n, T_m)
for (i in seq_len(n)) {
noise_i <- rnorm(T_m, sd = (1 / rsnr) * var(f1))
genetic_signal[i, ] <-
Geno[i, pos1] * f1 + Geno[i, pos2] * f2 + noise_i
covariate_signal[i, ] <-
Cov[i, 1L] * f1_cov + Cov[i, 2L] * f2_cov + Cov[i, 3L] * f3_cov
}
Y <- covariate_signal + genetic_signalThe covariate signal dominates the per-position scale of
Y, so without adjustment fSuSiE’s recovered effect curves
are biased toward the covariate-induced confounding rather than the true
genetic effect.
Account for covariates
mf_adjust_for_covariates(Y, Cov, method = "wavelet_eb")
fits a wavelet-domain empirical-Bayes regression of Y on
the covariates Cov and returns the residualised response
Y_adjusted = Y - Cov %*% fitted_func along with the fitted
covariate effect curves.
adj <- mf_adjust_for_covariates(Y, Cov)
str(adj, max.level = 1L)
#> List of 7
#> $ Y_adjusted : num [1:574, 1:128] 0.125 0.762 -0.284 0.222 -0.473 ...
#> $ X_adjusted : NULL
#> $ fitted_func: num [1:3, 1:128] 1.354 -0.268 -0.51 1.414 -0.41 ...
#> $ sigma2 : num 0.424
#> $ niter : int 5
#> $ converged : logi TRUE
#> $ method : chr "wavelet_eb"The first row of adj$fitted_func is the recovered effect
of the first covariate on the response curve; overlay it on the truth
f1_cov:
plot(adj$fitted_func[1L, ], type = "l", col = "blue",
lty = 2, lwd = 2,
xlab = "position", ylab = "covariate effect",
main = "Covariate 1: estimated vs true")
lines(f1_cov, lwd = 1.5)
legend("topright", lwd = c(1.5, 2), lty = c(1, 2),
legend = c("true f1_cov", "estimated"), bty = "n")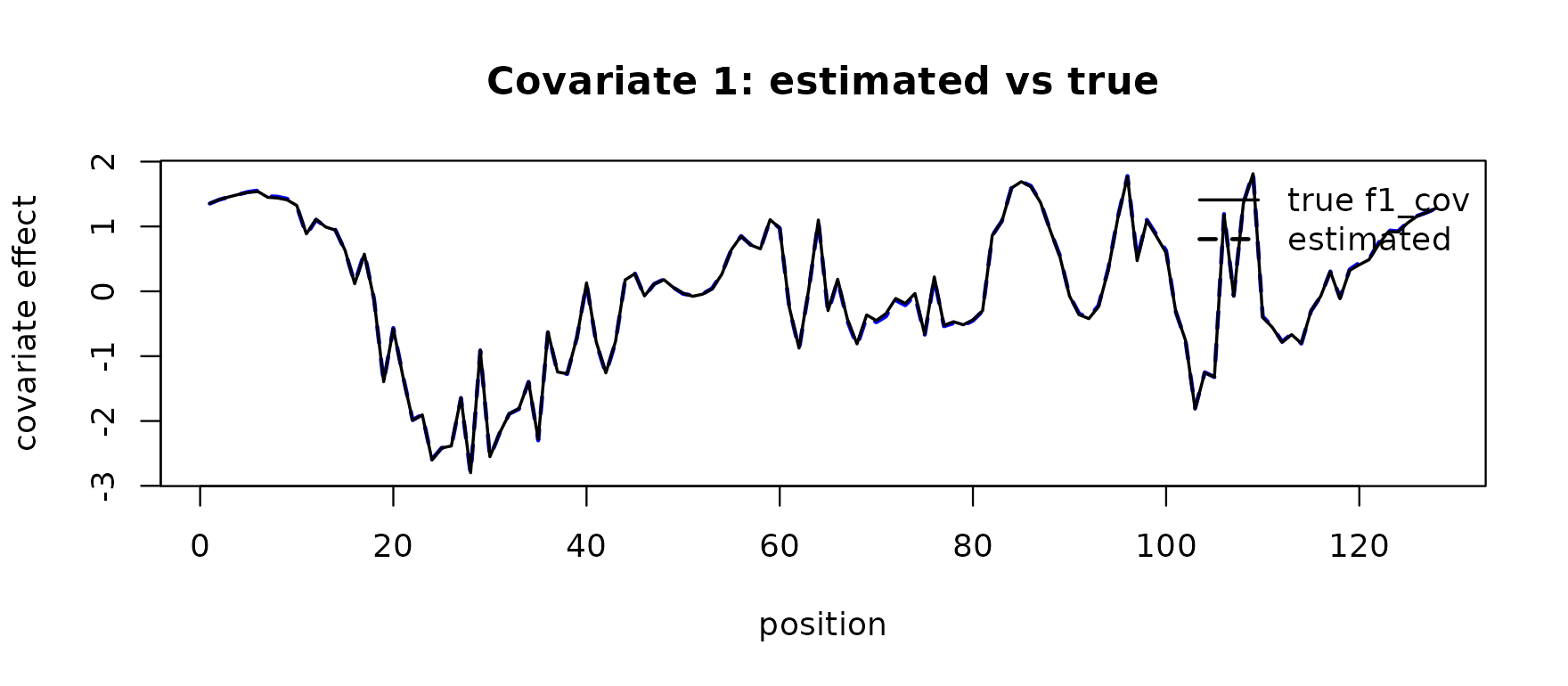
adj$Y_adjusted is the residualised response on the
original position grid, ready to feed into fsusie().
Recovering the genetic signal
A two-panel scatter shows that the adjusted response tracks the genetic-only signal far more tightly than the raw response:
par(mfrow = c(1L, 2L))
plot(Y, genetic_signal, pch = 19L, cex = 0.4,
xlab = "Observed Y", ylab = "Genetic signal",
main = "Before adjustment")
abline(0, 1, lty = 3, col = "grey50")
plot(adj$Y_adjusted, genetic_signal, pch = 19L, cex = 0.4,
xlab = "Y adjusted", ylab = "Genetic signal",
main = "After adjustment")
abline(0, 1, lty = 3, col = "grey50")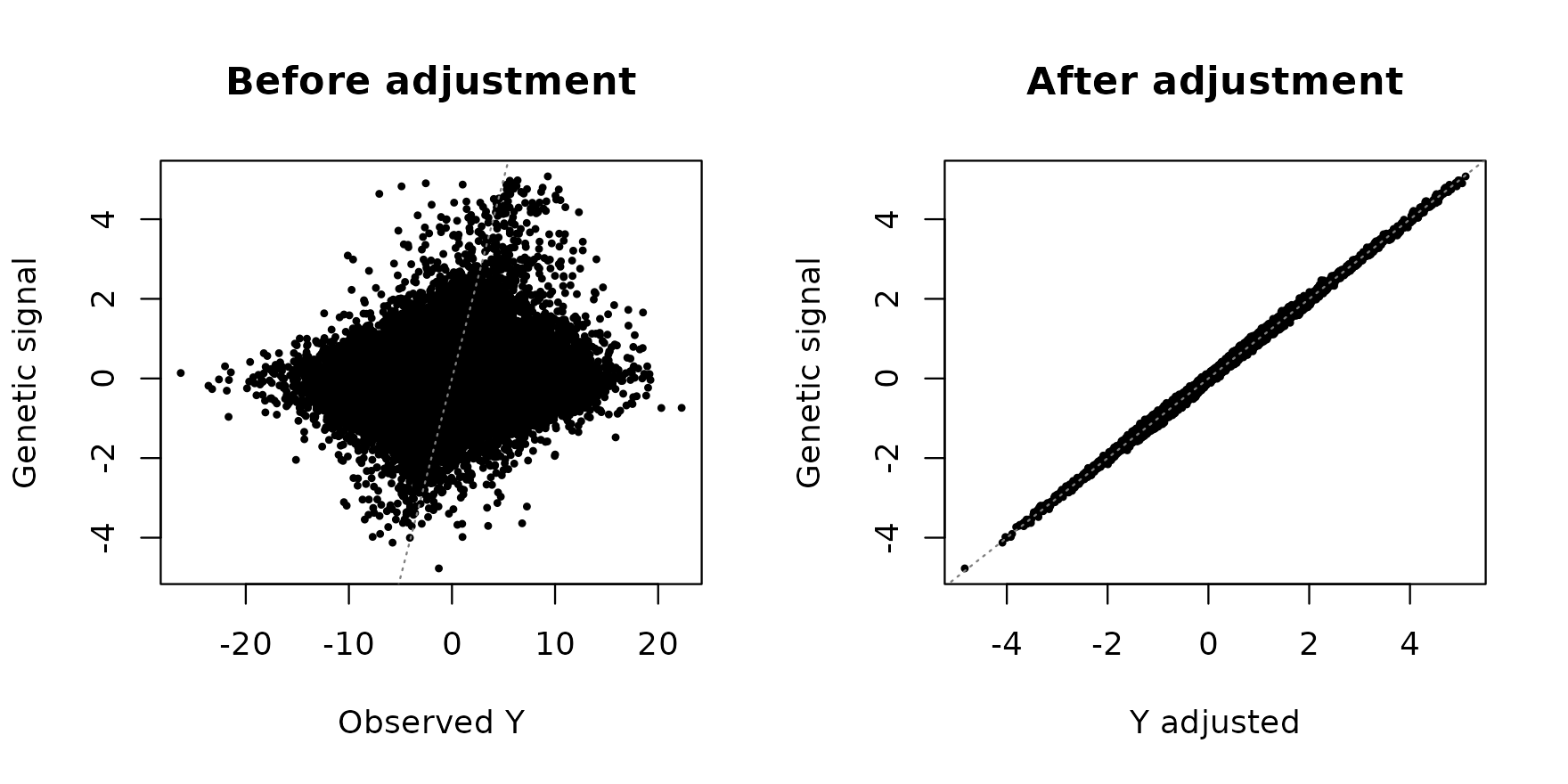
Fine-mapping the adjusted response
fit_adj <- fsusie(adj$Y_adjusted, Geno)
fit_adj$sets$cs
#> $L1
#> [1] 75 83
#>
#> $L2
#> [1] 25
fit_adj$pip[c(pos1, pos2)]
#> [1] 1.0000000 0.5001951
plot_dims <- mfsusie_plot_dimensions(fit_adj)
mfsusie_plot(fit_adj, effect_variables = c(pos1, pos2))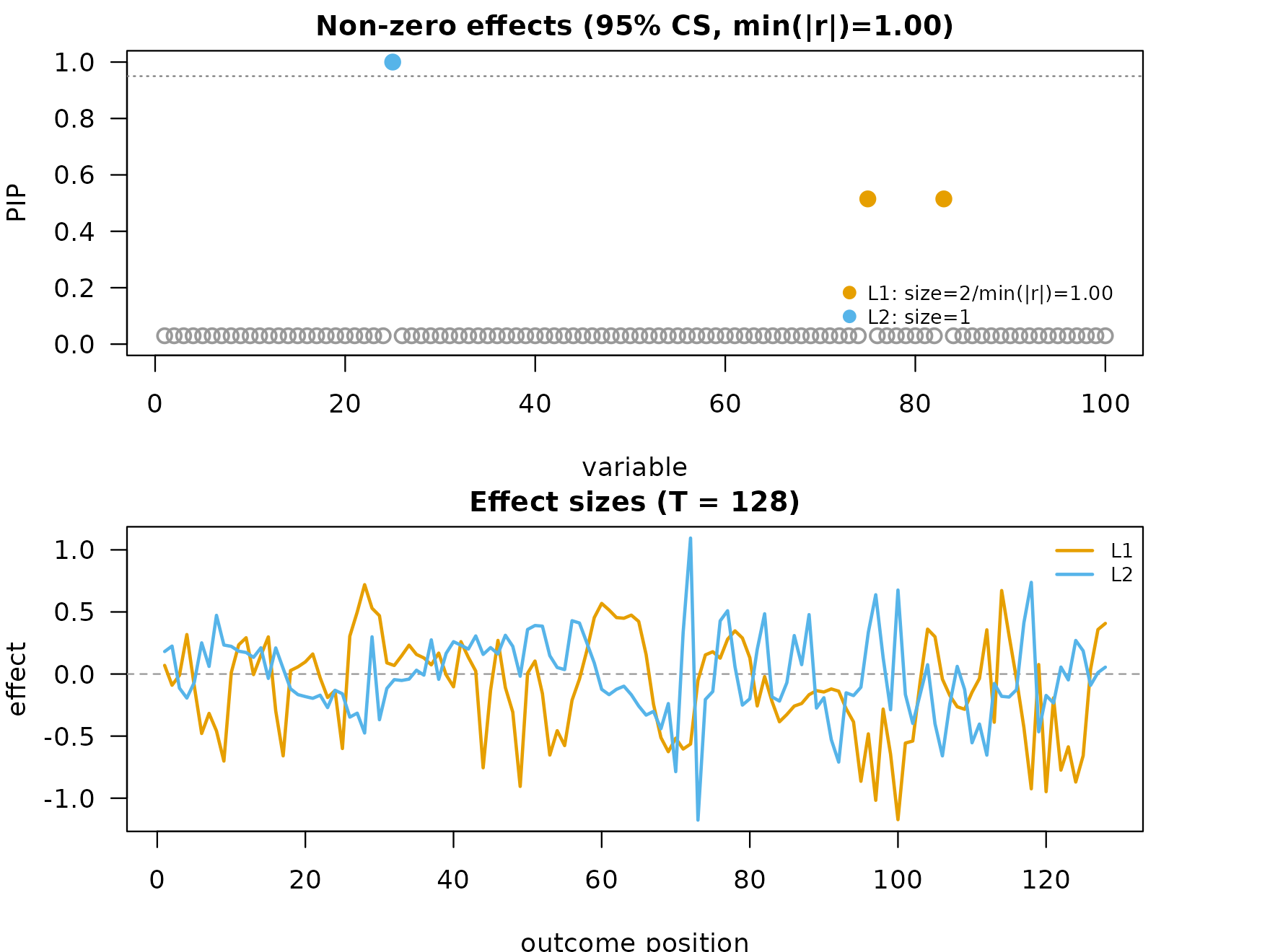
Comparison with no adjustment
For contrast, fit fsusie() directly on Y
without adjustment:
fit_unadj <- fsusie(Y, Geno)
fit_unadj$sets$cs
#> $L1
#> [1] 75 83
#>
#> $L3
#> [1] 48
#>
#> $L4
#> [1] 94
#>
#> $L5
#> [1] 97
#>
#> $L6
#> [1] 57
#>
#> $L7
#> [1] 60
#>
#> $L2
#> [1] 24 26
fit_unadj$pip[c(pos1, pos2)]
#> [1] 0.0001472286 0.5000000000fsusie() returns raw inverse-DWT effect curves via
coef(); position-space smoothing with a 95% credible band
is an opt-in post-processing step. Run
mf_post_smooth(fit, method = "TI") on each fit to get
translation-invariant cycle-spun curves, then read them back via
coef(fit_s, smooth_method = "TI"). Match each fitted CS to
its causal SNP by lead-PIP and overlay the smoothed recovered curve from
each fit against the ground-truth f1 and
f2.
fit_adj_s <- mf_post_smooth(fit_adj, method = "TI")
fit_unadj_s <- mf_post_smooth(fit_unadj, method = "TI")
cf_adj <- coef(fit_adj_s, smooth_method = "TI")[[1L]]
cf_unadj <- coef(fit_unadj_s, smooth_method = "TI")[[1L]]
match_cs <- function(fit, target_snp) {
if (length(fit$sets$cs) == 0L) return(NA_integer_)
leads <- vapply(fit$sets$cs, function(idx) {
idx[which.max(fit$pip[idx])]
}, integer(1L))
hit <- which(leads == target_snp)
if (length(hit) == 0L) NA_integer_ else hit[1L]
}
plot_effect <- function(snp, truth, col, label) {
l_adj <- match_cs(fit_adj, snp)
l_unadj <- match_cs(fit_unadj, snp)
rec_adj <- if (!is.na(l_adj)) cf_adj[l_adj, ] else NULL
rec_unadj <- if (!is.na(l_unadj)) cf_unadj[l_unadj, ] else NULL
yrng <- range(c(truth, rec_adj, rec_unadj), na.rm = TRUE)
plot(truth, pch = 20L, col = "grey30", cex = 0.7,
xlab = "position", ylab = "effect", ylim = yrng,
main = paste("Estimated effect:", label))
abline(h = 0, lty = 3, col = "grey50")
if (!is.null(rec_adj)) lines(rec_adj, col = col, lwd = 1.8)
if (!is.null(rec_unadj)) lines(rec_unadj, col = col, lwd = 1.5, lty = 2)
legend("topright",
legend = c("truth", "fSuSiE adjusted", "fSuSiE unadjusted"),
col = c("grey30", col, col),
lty = c(NA, 1, 2), lwd = c(NA, 1.8, 1.5),
pch = c(20, NA, NA), bty = "n", cex = 0.85)
}Estimated effect 1 (true causal at SNP 25):
plot_effect(pos1, f1, "blue", "f1 (SNP 25)")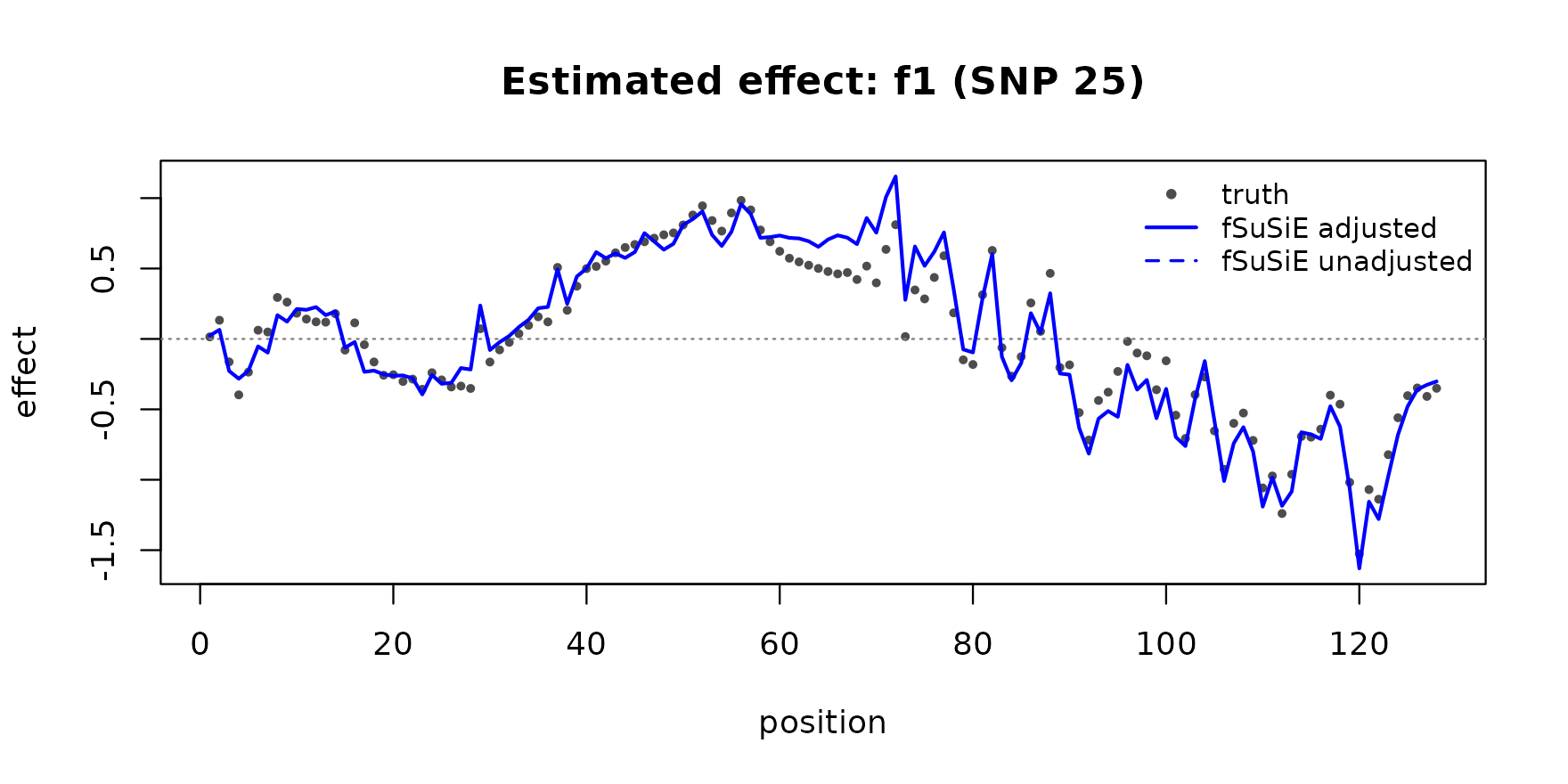
Estimated effect 2 (true causal at SNP 75):
plot_effect(pos2, f2, "darkgreen", "f2 (SNP 75)")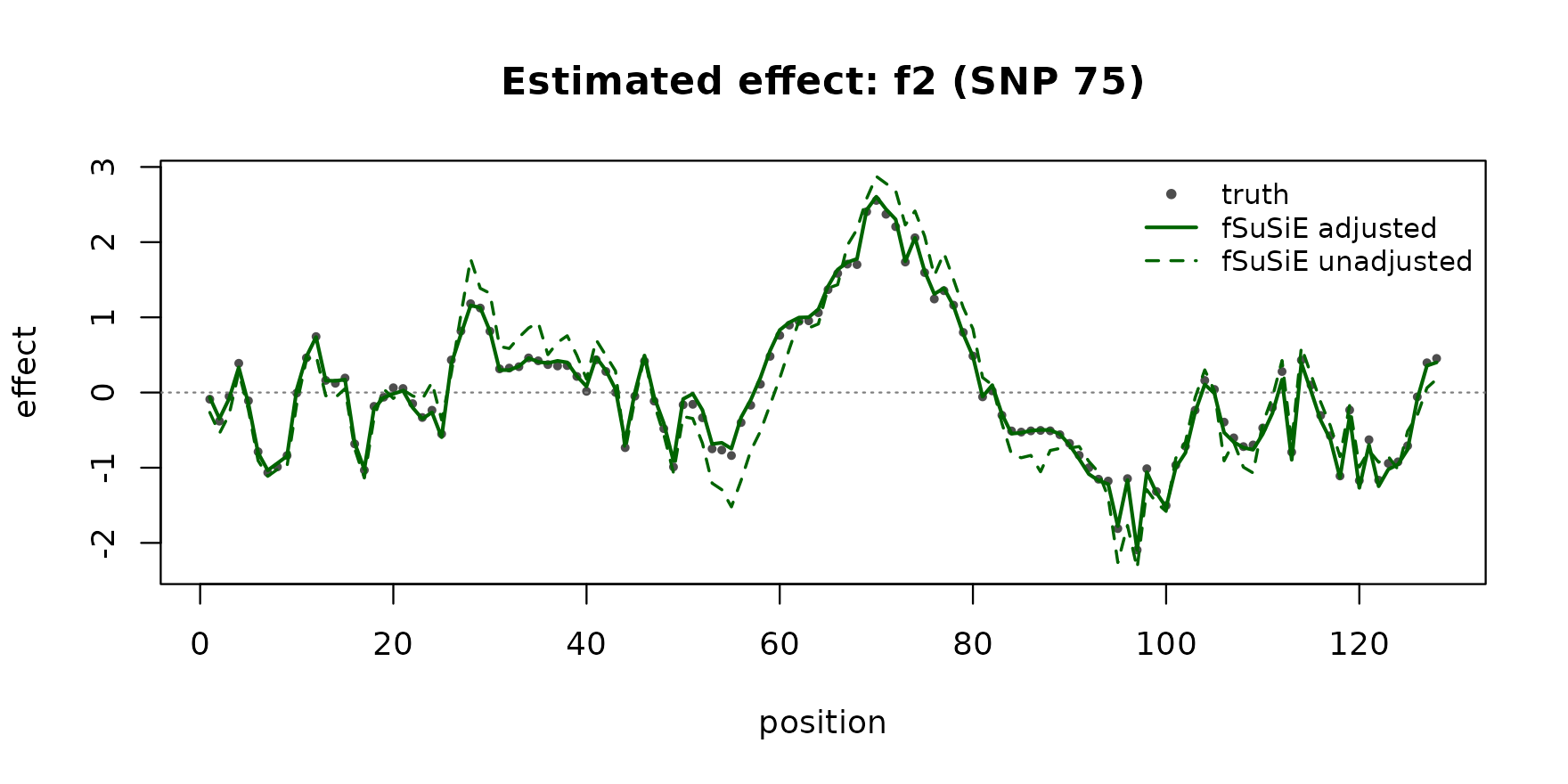
The TI-smoothed adjusted-fit curves track the truth on both signals at near-perfect correlation. The unadjusted fit is biased toward the covariate-induced confounding and the recovered curves drift away from the truth.
Scalar traits: OLS adjustment with the FWL correction
When the response is a scalar (T = 1) or smoothness
across positions is irrelevant, the closed-form OLS residualization is
the right tool. Pass method = "ols" to use it. When the
covariates are correlated with genotype, also pass X so the
function returns the Frisch-Waugh-Lovell-corrected
X_adjusted:
# Scalar response derived from the same covariate-confounded
# data: take the per-individual mean of Y as the scalar trait
# and wrap it as a single-column matrix.
Y_scalar <- matrix(rowMeans(Y), ncol = 1L)
adj_ols <- mf_adjust_for_covariates(Y_scalar, Cov, X = Geno,
method = "ols")
str(adj_ols, max.level = 1L)
#> List of 4
#> $ Y_adjusted : num [1:574, 1] -0.00905 0.03383 -0.04059 0.03828 0.00103 ...
#> $ X_adjusted : num [1:574, 1:100] -0.0171 -0.0109 -0.0104 -0.0107 -0.0458 ...
#> $ fitted_func: num [1:3, 1] -3.88e-05 -4.15e-04 -5.67e-04
#> $ method : chr "ols"adj_ols returns:
-
Y_adjusted: the scalar response after projecting out the covariate column space. -
X_adjusted: the genotype matrix after the same projection (the FWL correction). WhenCovis correlated withGeno, usingX_adjustedinstead of the originalGenoremoves the bias in the downstream fit.
A scalar susie() fit on the OLS-adjusted data:
fit_scalar <- susieR::susie(adj_ols$X_adjusted,
as.numeric(adj_ols$Y_adjusted),
L = 5L, verbose = FALSE)
fit_scalar$sets$cs
#> NULL
fit_scalar$pip[c(pos1, pos2)]
#> [1] 0 0For T > 1 use method = "wavelet_eb" (the
default demonstrated above). Use method = "ols" for scalar
traits or when smooth covariate effects across positions are not
expected.
Next steps
For colocalization across two functional fits, see the colocalization vignette. For post-fit smoothing of effect curves, see the post-processing vignette.
Session info
This is the version of R and the packages that were used to generate these results.
sessionInfo()
#> R version 4.4.3 (2025-02-28)
#> Platform: x86_64-conda-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS/LAPACK: /home/runner/work/mfsusieR/mfsusieR/.pixi/envs/r44/lib/libopenblasp-r0.3.32.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: Etc/UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] wavethresh_4.7.3 MASS_7.3-65 susieR_0.16.1 mfsusieR_0.0.2
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 generics_0.1.4 ashr_2.2-63
#> [4] lattice_0.22-9 digest_0.6.39 magrittr_2.0.5
#> [7] evaluate_1.0.5 grid_4.4.3 RColorBrewer_1.1-3
#> [10] fastmap_1.2.0 plyr_1.8.9 jsonlite_2.0.0
#> [13] Matrix_1.7-5 reshape_0.8.10 mixsqp_0.3-54
#> [16] scales_1.4.0 truncnorm_1.0-9 invgamma_1.2
#> [19] textshaping_1.0.5 jquerylib_0.1.4 cli_3.6.6
#> [22] crayon_1.5.3 zigg_0.0.2 rlang_1.2.0
#> [25] deconvolveR_1.2-1 LaplacesDemon_16.1.8 splines_4.4.3
#> [28] cachem_1.1.0 yaml_2.3.12 otel_0.2.0
#> [31] ebnm_1.0-55 tools_4.4.3 SQUAREM_2026.1
#> [34] parallel_4.4.3 dplyr_1.2.1 ggplot2_4.0.3
#> [37] Rfast_2.1.5.2 vctrs_0.7.3 R6_2.6.1
#> [40] matrixStats_1.5.0 lifecycle_1.0.5 fs_2.1.0
#> [43] htmlwidgets_1.6.4 trust_0.1-9 ragg_1.5.2
#> [46] irlba_2.3.7 pkgconfig_2.0.3 desc_1.4.3
#> [49] RcppParallel_5.1.11-2 pkgdown_2.2.0 bslib_0.10.0
#> [52] pillar_1.11.1 gtable_0.3.6 glue_1.8.1
#> [55] Rcpp_1.1.1-1.1 systemfonts_1.3.2 xfun_0.57
#> [58] tibble_3.3.1 tidyselect_1.2.1 dichromat_2.0-0.1
#> [61] knitr_1.51 farver_2.1.2 htmltools_0.5.9
#> [64] rmarkdown_2.31 compiler_4.4.3 S7_0.2.2
#> [67] horseshoe_0.2.0