Joint multi-outcome fine-mapping
Gao Wang, Anjing Liu and William Denault
Source:vignettes/mfsusie_intro.Rmd
mfsusie_intro.Rmdmfsusie() extends fsusie() from one
functional response to many. Each outcome m is a matrix
n x T_m; outcomes are typically functional (DNAm tracks,
RNA-seq coverage, ATAC-seq profiles) and T_m may differ
across outcomes. The fit returns one credible set list and one PIP
vector per variable, combining evidence from every outcome. Scalar
outcomes (T_m = 1) are supported through the same interface
but susieR::susie() is typically the better tool for that
case; the strength of mfsusie() is joint fine-mapping
across multiple functional layers.
Multi-outcome example
multiomic_example is a four-outcome simulated cis-QTL
example. We use the two functional outcomes here: DNAm with
T = 64 irregular CpGs and RNA-seq with T = 32
exon-body positions. Two causal SNPs are shared across both outcomes;
per-outcome shapes and signs differ. Genotypes use the
susieR::N3finemapping LD scaffold for realistic
correlation.
Fit
fit <- mfsusie(multiomic_example$X, Y_list, pos = pos_list)
#> HINT: ncol(Y) is not 2^J or positions are unevenly spaced; interpolated to a regular dyadic grid.
#> iter ELBO delta sigma2 mem V extras
#> 1 -77313.8971 - [0.998, 0.999, 1.000] 0.16 GB [2.38e-02, 1.41e-02, 0 x 3] pi_null=[0.78, 1.00]
#> iter 2: max|d(alpha,PIP)|=6.94e-03, V=[2.55e-02, 1.41e-02, 0 x 3] [mem: 0.16 GB]
#> iter 3: max|d(alpha,PIP)|=7.24e-05, V=[2.55e-02, 1.41e-02, 0 x 3] -- converged (alpha_pip_fixed_point) [mem: 0.16 GB]
#> [L_greedy] 1 round, greedy_lbf_cutoff=0.100, final L=5
#> round L min(lbf) action
#> 1 5 0.000 saturated
fit$niter; fit$converged
#> [1] 3
#> [1] TRUE
length(fit$sets$cs)
#> [1] 2
fit$pip[multiomic_example$causal_snps]
#> [1] 1.0000000 0.9961233Post-hoc per-outcome causal-configuration assessment
For each credible set, ask which outcomes share the causal variant.
susie_post_outcome_configuration(fit, by = "outcome", method = ...)
runs one of two post-hoc analyses on the joint mfsusie fit; call again
with the other method to get both:
-
method = "susiex"(default): combinatorial enumeration. Posterior probability of each of the2^Mbinary “which-outcomes-share-the-causal” patterns, plus a per-outcome marginal probability of being active. One list element per CS index shared across all outcomes, so forMoutcomes sharingKCSes,length(configs$susiex) == K. -
method = "coloc_pairwise": standard H0/H1/H2/H3/H4 coloc posteriors for every (outcome pair, CS pair) (one data.frame).
summary() tidies either result into a colour-coded
table: green for active / shared, yellow for ambiguous, dim for
below-threshold. By default it filters to rows that show signal (pass
signal_only = FALSE to see everything).
configs <- susieR::susie_post_outcome_configuration(fit, by = "outcome",
method = "susiex")
summary(configs)#>
#> SuSiEx: per-trait marginal P(active) per CS tuple
#> prob_thresh = 0.80, ambiguous = [0.50, 0.80)
#>
#> CS tuple trait_1_outcome_1 trait_1_outcome_2 top pattern top P
#> -------- ----------------- ----------------- ----------- -----
#> (1,1) 1.000 1.000 11 1.000
#> (2,2) 1.000 1.000 11 1.000For the coloc_pairwise analysis the table has one row
per (outcome pair) x (CS pair), which on a 4-outcome fit with 3 shared
CSes means choose(4, 2) * 3 * 3 = 54 rows. We pass
signal_only = TRUE (the default) so H0-dominant rows drop
out and we see only the pairs that carry colocalisation evidence:
pairwise <- susieR::susie_post_outcome_configuration(fit, by = "outcome",
method = "coloc_pairwise")
summary(pairwise)#>
#> Coloc pairwise: dominant hypothesis per (trait, trait', l1, l2)
#> H0 no signal | H1 trait1-only | H2 trait2-only | H3 distinct | H4 shared
#>
#> trait1 trait2 l1 l2 hit1 hit2 PP.H0 PP.H1 PP.H2 PP.H3 PP.H4 verdict
#> ----------------- ----------------- -- -- ------ ------ ----- ----- ----- ----- ----- -----------
#> trait_1_outcome_1 trait_1_outcome_2 1 1 snp_97 snp_97 0.000 0.000 0.000 0.000 1.000 H4 shared
#> trait_1_outcome_1 trait_1_outcome_2 1 2 snp_97 snp_42 0.000 0.000 0.000 1.000 0.000 H3 distinct
#> trait_1_outcome_1 trait_1_outcome_2 2 1 snp_42 snp_97 0.000 0.000 0.000 1.000 0.000 H3 distinct
#> trait_1_outcome_1 trait_1_outcome_2 2 2 snp_42 snp_42 0.000 0.000 0.000 0.000 1.000 H4 sharedIndex the raw objects directly when you need full per-CS detail:
configs$susiex[[1L]]$marginal_prob # per-outcome marginal P(active)
#> trait_1_outcome_1 trait_1_outcome_2
#> 1 1
configs$susiex[[1L]]$config_prob # 2^M configuration probabilities
#> [1] 2.905561e-277 1.253220e-121 2.318476e-156 1.000000e+00
head(pairwise$coloc_pairwise) # raw coloc data.frame
#> trait1 trait2 l1 l2 hit1 hit2 PP.H0
#> 1 trait_1_outcome_1 trait_1_outcome_2 1 1 snp_97 snp_97 5.662559e-272
#> 2 trait_1_outcome_1 trait_1_outcome_2 1 2 snp_97 snp_42 2.093928e-196
#> 3 trait_1_outcome_1 trait_1_outcome_2 2 1 snp_42 snp_97 2.776337e-206
#> 4 trait_1_outcome_1 trait_1_outcome_2 2 2 snp_42 snp_42 4.929588e-136
#> PP.H1 PP.H2 PP.H3 PP.H4
#> 1 2.549304e-120 5.332741e-155 0.0004016223 9.995984e-01
#> 2 9.426937e-45 2.221217e-152 1.0000000000 2.649112e-47
#> 3 1.061848e-117 2.614629e-89 1.0000000000 2.414078e-95
#> 4 1.885387e-47 5.229257e-92 0.0000000000 1.000000e+00Inspecting the fit
mfsusie_plot() shows everything in one call: PIPs in the
top-left tile (CS-colored), then one effect-curve panel per outcome with
TI-smoothed curves, 95% credible bands, and affected-region bars at the
bottom of each panel.
fit_s <- mf_post_smooth(fit)When the fit has both M > 1 outcomes and several
credible sets, facet_cs = "stack" lays out one effect panel
per (outcome, CS) pair stacked vertically below the PIP
panel, which keeps the per-CS curves at full width. The default
facet_cs = "auto" falls back to a per-outcome overlay grid
when M > 1; switch to "stack" when you want
a separated view per CS.
mfsusie_plot_dimensions() returns a
(width, height) recommendation in inches sized to the
number of panels the fit will produce. Pass it through to the chunk so
each cell keeps a legible amount of vertical space.
plot_dims <- mfsusie_plot_dimensions(fit_s, facet_cs = "stack")
plot_dims
#> $width
#> [1] 8
#>
#> $height
#> [1] 15
#>
#> $n_cells
#> [1] 5
mfsusie_plot(fit_s, facet_cs = "stack",
effect_variables = multiomic_example$causal_snps)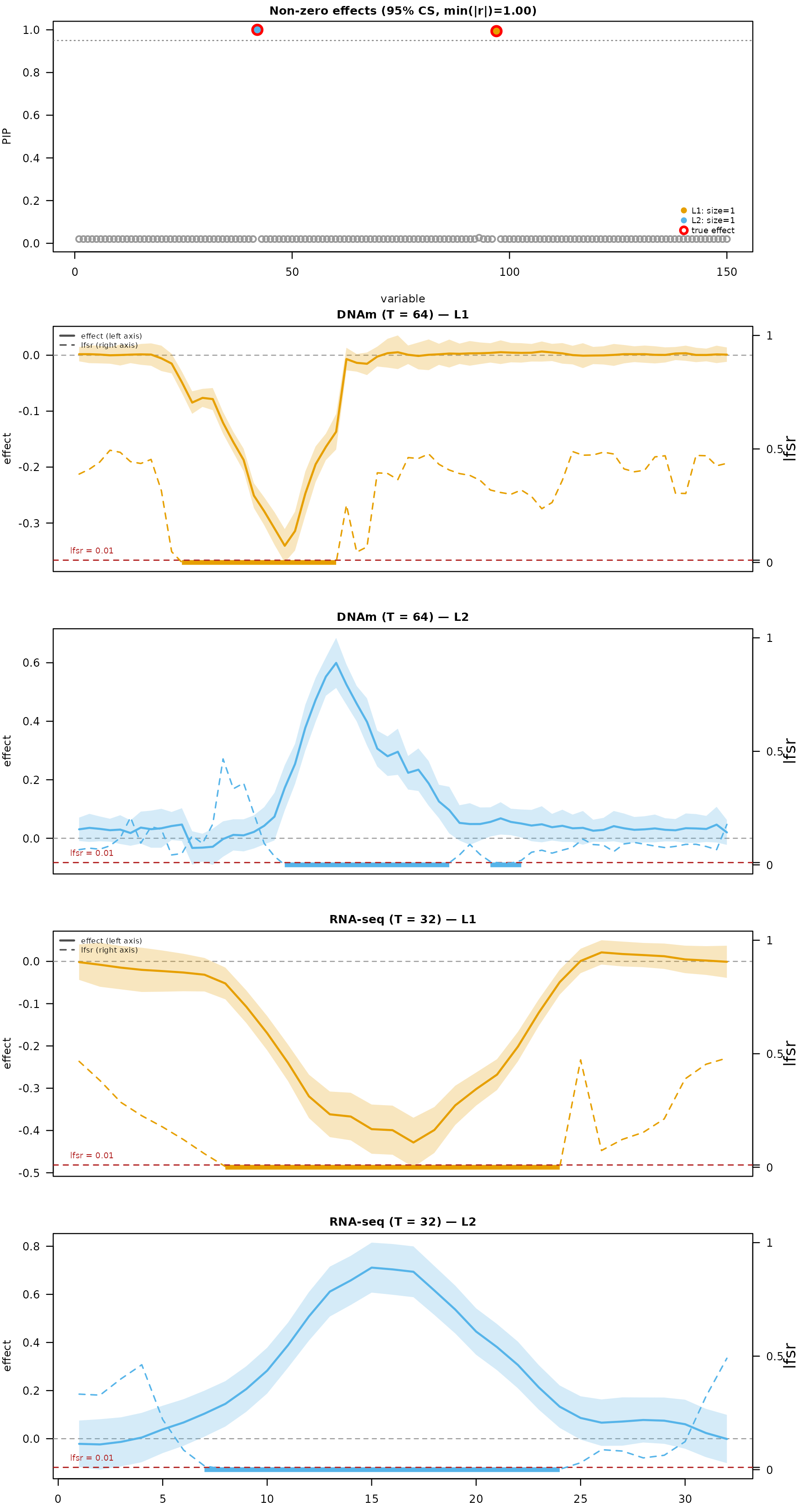
fit$sets$cs
#> $L1
#> [1] 97
#>
#> $L2
#> [1] 42coef(fit) returns a list of length M.
Element m is an L x T_m matrix containing the
per-effect curve for outcome m.
mf_summarize_effects(fit) returns a tidy data frame of
the per-(CS, outcome) credibly-non-zero windows (start, end,
n_positions), derived from the credible bands of the picked smoother.
Useful when you want a tabular handle on which positions each CS lights
up at, separately from the plot.
mf_summarize_effects(fit_s)
#> cs_index outcome start end n_positions
#> 1 1 1 11 26 16
#> 2 2 1 19 37 19
#> 3 2 1 41 44 4
#> 4 1 2 8 24 17
#> 5 2 2 7 24 18Choosing prior_variance_scope
-
"per_outcome"(default; used above): one mixture-of-normals prior shared across every wavelet scale of an outcome. Appropriate when scales are exchangeable a priori, and roughlyS_m-fold cheaper per IBSS iter (one mixsqp call per outcome instead of one per (outcome, scale)). -
"per_scale": one mixture per (outcome, scale), letting different scales adapt independently. More flexible at the cost of anS_m-fold larger M-step; useful when shape varies systematically with scale.
Switch between them by changing one argument; the rest of the API and the fit object structure are identical.
Wavelet-domain preprocessing
Three arguments control preprocessing of the per-outcome wavelet matrix before the IBSS loop:
-
wavelet_magnitude_cutoff(default0): wavelet columns withmedian(|column|) <= wavelet_magnitude_cutoffare masked, contributing(Bhat = 0, Shat = 1)to the SER step. Use a small positive value when the response has sparse non-zero entries (e.g., ATAC-seq read pileups). -
wavelet_standardize(defaultTRUE): centers and scales each packed wavelet coefficient column before IBSS. This is a linear, invertible rescaling; fitted values and coefficients are mapped back to the original response scale. -
wavelet_qnorm(defaultFALSE): applies a column-wise rank-normal transform to the wavelet matrix so each wavelet position has marginalN(0, 1)across samples by construction. Useful when the per-outcome residuals are heavy-tailed (read counts, sparse pileups). The transform is non-linear, socoef(fit)curves recover shape and CS membership but not amplitude on the rawYscale.
wavelet_magnitude_cutoff also routes through
mf_adjust_for_covariates(). The qnorm and
wavelet-standardize options are fine-mapping preprocessing choices.
When to reach for mfSuSiE over fSuSiE plus colocalization
Use mfsusie() when (i) a variable plausibly affects
multiple distinct phenotypes (different tissues, different molecular
layers); (ii) the phenotypes have heterogeneous shapes, since mfSuSiE
does not require a shared T (some scalar, some functional,
some at different sampling resolutions are fine in the same fit); (iii)
a single credible-set list combining all the evidence is preferable to a
post-hoc colocalization across separate fsusie fits.
Multi-trait data with missing samples
Multi-tissue and multi-cell-type studies often leave some samples
unprofiled for some outcomes. Set the corresponding rows of
Y[[m]] to NA before calling
mfsusie(); the package restricts every regression and
variance update to the observed rows for that outcome. Both
wavelet_qnorm = FALSE (default) and
wavelet_qnorm = TRUE handle missing rows correctly.
set.seed(42)
n <- 80; p <- 60; T_m <- 8; M <- 3
X_miss <- matrix(rbinom(n * p, 2, 0.3), n, p)
X_miss <- scale(X_miss)
# Plant one causal SNP shared across all three traits.
causal_j <- 5L
signal <- as.numeric(X_miss[, causal_j]) * 2
Y_miss <- lapply(seq_len(M), function(m)
matrix(signal, n, T_m) + matrix(rnorm(n * T_m, sd = 0.5), n, T_m))
# Trait 2 is missing 20 % of rows; trait 3 is missing 30 %.
Y_miss[[2]][sample(n, 16), ] <- NA
Y_miss[[3]][sample(n, 24), ] <- NA
# Per-trait observed sample sizes after masking.
sapply(Y_miss, function(y) sum(complete.cases(y)))
#> [1] 80 64 56
wavelet_qnorm = FALSE
t_noq <- system.time(
fit_noq <- mfsusie(X_miss, Y_miss,
wavelet_qnorm = FALSE)
)
#> iter ELBO delta sigma2 mem V extras
#> 1 -2181.4266 - [0.987, 0.997, 1.000] 0.16 GB [1.20e-01, 0 x 4] pi_null=[0.87, 1.00]
#> iter 2: max|d(alpha,PIP)|=2.71e-36, V=[1.19e-01, 0 x 4] -- converged (alpha_pip_fixed_point) [mem: 0.16 GB]
#> [L_greedy] 1 round, greedy_lbf_cutoff=0.100, final L=5
#> round L min(lbf) action
#> 1 5 0.000 saturated
cat(sprintf("Runtime: %.1f s niter: %d CS: %d\n",
t_noq["elapsed"], fit_noq$niter, length(fit_noq$sets$cs)))
#> Runtime: 0.7 s niter: 2 CS: 1
fit_noq$pip[causal_j] # causal PIP
#> [1] 1
wavelet_qnorm = TRUE
t_q <- system.time(
fit_q <- mfsusie(X_miss, Y_miss,
wavelet_qnorm = TRUE)
)
#> iter ELBO delta sigma2 mem V extras
#> 1 -2091.2739 - [0.987, 0.994, 0.997] 0.16 GB [5.28e-02, 0 x 4] pi_null=[0.87, 1.00]
#> iter 2: max|d(alpha,PIP)|=2.22e-16, V=[5.15e-02, 0 x 4] -- converged (alpha_pip_fixed_point) [mem: 0.16 GB]
#> [L_greedy] 1 round, greedy_lbf_cutoff=0.100, final L=5
#> round L min(lbf) action
#> 1 5 0.000 saturated
cat(sprintf("Runtime: %.1f s niter: %d CS: %d\n",
t_q["elapsed"], fit_q$niter, length(fit_q$sets$cs)))
#> Runtime: 0.7 s niter: 2 CS: 1
fit_q$pip[causal_j]
#> [1] 1Runtime summary
data.frame(
path = c("qnorm=FALSE", "qnorm=TRUE"),
runtime = c(t_noq["elapsed"], t_q["elapsed"]),
niter = c(fit_noq$niter, fit_q$niter),
n_cs = c(length(fit_noq$sets$cs), length(fit_q$sets$cs)),
causal_pip = c(fit_noq$pip[causal_j], fit_q$pip[causal_j])
)
#> path runtime niter n_cs causal_pip
#> 1 qnorm=FALSE 0.712 2 1 1
#> 2 qnorm=TRUE 0.702 2 1 1Session info
This is the version of R and the packages that were used to generate these results.
sessionInfo()
#> R version 4.4.3 (2025-02-28)
#> Platform: x86_64-conda-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS/LAPACK: /home/runner/work/mfsusieR/mfsusieR/.pixi/envs/r44/lib/libopenblasp-r0.3.32.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: Etc/UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] susieR_0.16.1 mfsusieR_0.0.2
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 generics_0.1.4 ashr_2.2-63
#> [4] lattice_0.22-9 digest_0.6.39 magrittr_2.0.5
#> [7] RColorBrewer_1.1-3 evaluate_1.0.5 grid_4.4.3
#> [10] fastmap_1.2.0 plyr_1.8.9 jsonlite_2.0.0
#> [13] Matrix_1.7-5 reshape_0.8.10 mixsqp_0.3-54
#> [16] fansi_1.0.7 scales_1.4.0 truncnorm_1.0-9
#> [19] invgamma_1.2 textshaping_1.0.5 jquerylib_0.1.4
#> [22] cli_3.6.6 crayon_1.5.3 zigg_0.0.2
#> [25] rlang_1.2.0 deconvolveR_1.2-1 LaplacesDemon_16.1.8
#> [28] splines_4.4.3 cachem_1.1.0 yaml_2.3.12
#> [31] otel_0.2.0 ebnm_1.0-55 tools_4.4.3
#> [34] SQUAREM_2026.1 parallel_4.4.3 dplyr_1.2.1
#> [37] wavethresh_4.7.3 ggplot2_4.0.3 Rfast_2.1.5.2
#> [40] vctrs_0.7.3 R6_2.6.1 matrixStats_1.5.0
#> [43] lifecycle_1.0.5 fs_2.1.0 htmlwidgets_1.6.4
#> [46] MASS_7.3-65 trust_0.1-9 ragg_1.5.2
#> [49] irlba_2.3.7 pkgconfig_2.0.3 desc_1.4.3
#> [52] RcppParallel_5.1.11-2 pkgdown_2.2.0 bslib_0.10.0
#> [55] pillar_1.11.1 gtable_0.3.6 glue_1.8.1
#> [58] Rcpp_1.1.1-1.1 systemfonts_1.3.2 xfun_0.57
#> [61] tibble_3.3.1 tidyselect_1.2.1 dichromat_2.0-0.1
#> [64] knitr_1.51 farver_2.1.2 htmltools_0.5.9
#> [67] rmarkdown_2.31 compiler_4.4.3 S7_0.2.2
#> [70] horseshoe_0.2.0