
RNA-seq expression#
Miniprotocol Timing#
Timing <3.5 hours
Overview#
This miniprotocol shows the use of various modules to prepare reference data, perform RNA-seq calling, quantify expression, conduct quality control and normalize data. The modules are as follows:
RNA_calling.ipynb(steps i-v): Quantifying expression from RNA-seq databulk_expression_QC.ipynb(step vi): Sample level RNA-seq quality controlbulk_expression_normalization.ipynb(step vii): Bulk RNA-seq counts normalization
Steps#
i. Perform data quality summary via fastqc#
sos run pipeline/RNA_calling.ipynb fastqc \
--cwd output/rnaseq/fastqc \
--sample-list data/fastq.list.txt \
--data-dir data/fastq
ii. Cut adaptor (Optional)#
sos run pipeline/RNA_calling.ipynb fastp_trim_adaptor \
--cwd output/rnaseq --sample-list data/fastq.list.txt \
--data-dir data/fastq --STAR-index reference_data/STAR_Index/ \
--gtf reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.ERCC.gtf \
--reference-fasta reference_data/GRCh38_full_analysis_set_plus_decoy_hla.noALT_noHLA_noDecoy_ERCC.fasta \
--ref-flat reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.ERCC.ref.flat
iii. Read alignment via STAR and QC via Picard#
sos run pipeline/RNA_calling.ipynb STAR_align \
--cwd output/rnaseq/bam --sample-list data/fastq.list.txt \
--data-dir data/fastq --STAR-index reference_data/STAR_Index/ \
--gtf reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.ERCC.gtf \
--reference-fasta reference_data/GRCh38_full_analysis_set_plus_decoy_hla.noALT_noHLA_noDecoy_ERCC.fasta \
--ref-flat reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.ERCC.ref.flat \
--chimSegmentMin 0 \
-J 50 --mem 200G --numThreads 8
iv. Call gene-level RNA expression via rnaseqc#
sos run pipeline/RNA_calling.ipynb rnaseqc_call \
--cwd data/bam \
--sample-list data/fastq.list.txt \
--data-dir data/fastq \
--gtf reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.collapse_only.gene.gtf \
--reference-fasta reference_data/GRCh38_full_analysis_set_plus_decoy_hla.noALT_noHLA_noDecoy_ERCC.fasta \
--varVCFfile reference_data/ZOD14598_AD_GRM_WGS_2021-04-29_all.recalibrated_variants.leftnorm.filtered.AF.WASP.vcf \
--bam_list data/bam/sample_bam_list.txt
v. Call transcript level RNA expression via RSEM#
sos run pipeline/RNA_calling.ipynb rsem_call \
--cwd data/bam \
--sample-list data/fastq.list.txt \
--data-dir data/fastq \
--STAR-index reference_data/STAR_Index/ \
--gtf reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.ERCC.gtf \
--reference-fasta reference_data/GRCh38_full_analysis_set_plus_decoy_hla.noALT_noHLA_noDecoy_ERCC.fasta \
--ref-flat reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.ERCC.ref.flat \
--varVCFfile reference_data/ZOD14598_AD_GRM_WGS_2021-04-29_all.recalibrated_variants.leftnorm.filtered.AF.WASP.vcf \
--bam_list data/bam/sample_bam_list.txt \
--RSEM-index reference_data/RSEM_Index
vi. Multi-sample RNA-seq QC#
sos run pipeline/bulk_expression_QC.ipynb qc \
--cwd output/rnaseq \
--tpm-gct data/rnaseq/bulk_rnaseq_tmp_matrix.bed \
--counts-gct data/rnaseq/bulk_rnaseq_count_matrix.bed
vii. Multi-sample read count normalization#
sos run pipeline/bulk_expression_normalization.ipynb normalize \
--cwd output/rnaseq \
--tpm-gct output/rnaseq/bulk_rnaseq_tmp_matrix.low_expression_filtered.outlier_removed.tpm.gct.gz \
--counts-gct output/rnaseq/bulk_rnaseq_tmp_matrix.low_expression_filtered.outlier_removed.geneCount.gct.gz \
--annotation-gtf reference_data/Homo_sapiens.GRCh38.103.chr.reformatted.collapse_only.gene.ERCC.gtf \
--count-threshold 1 --sample_participant_lookup data/rnaseq/sample_participant_lookup.txt
Anticipated Results#
The final output contained QCed and normalized expression data in a bed.gz file. This file is ready for use in TensorQTL.
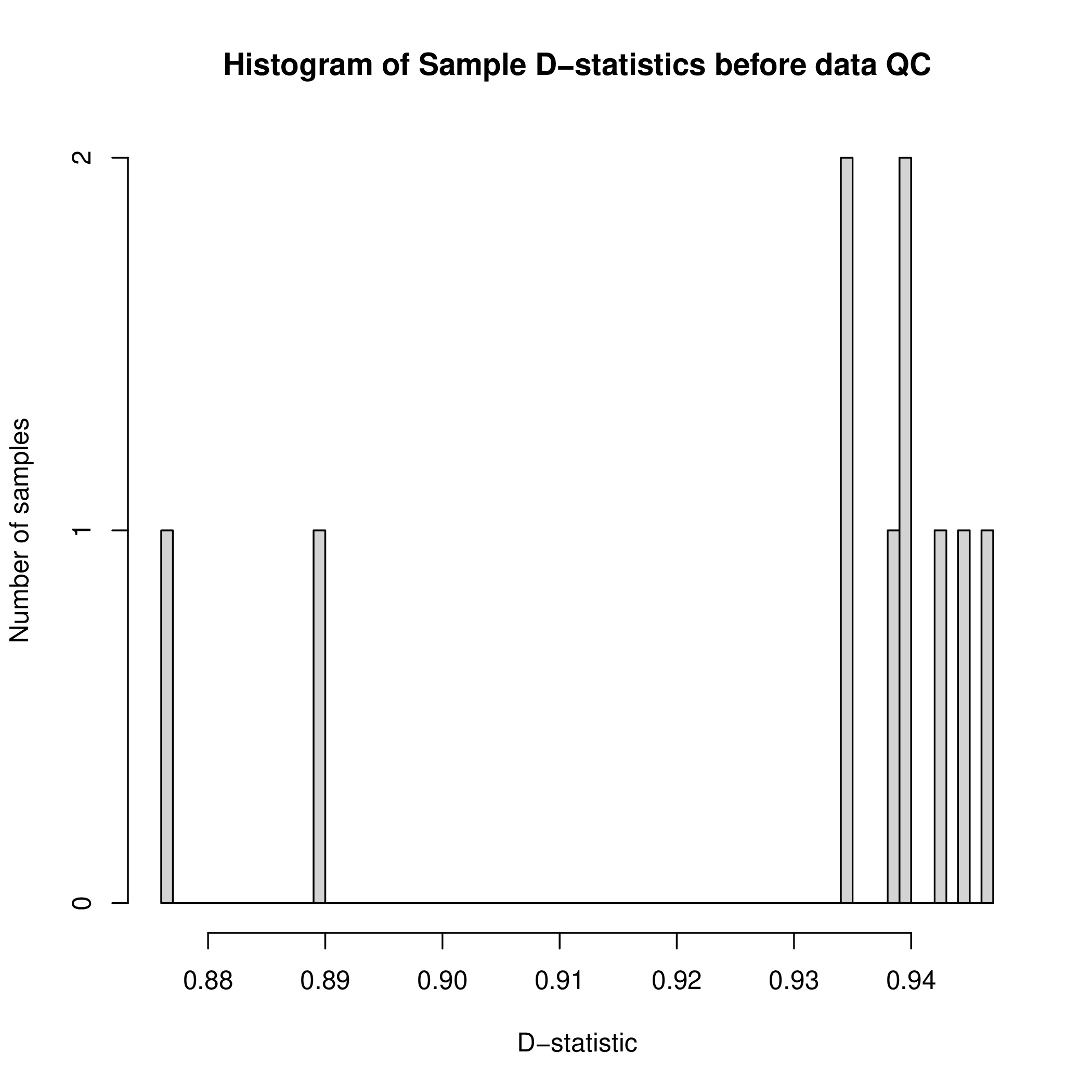
Figure 1A. Bulk RNA-Seq Quality Control D-Statistic Distribution.
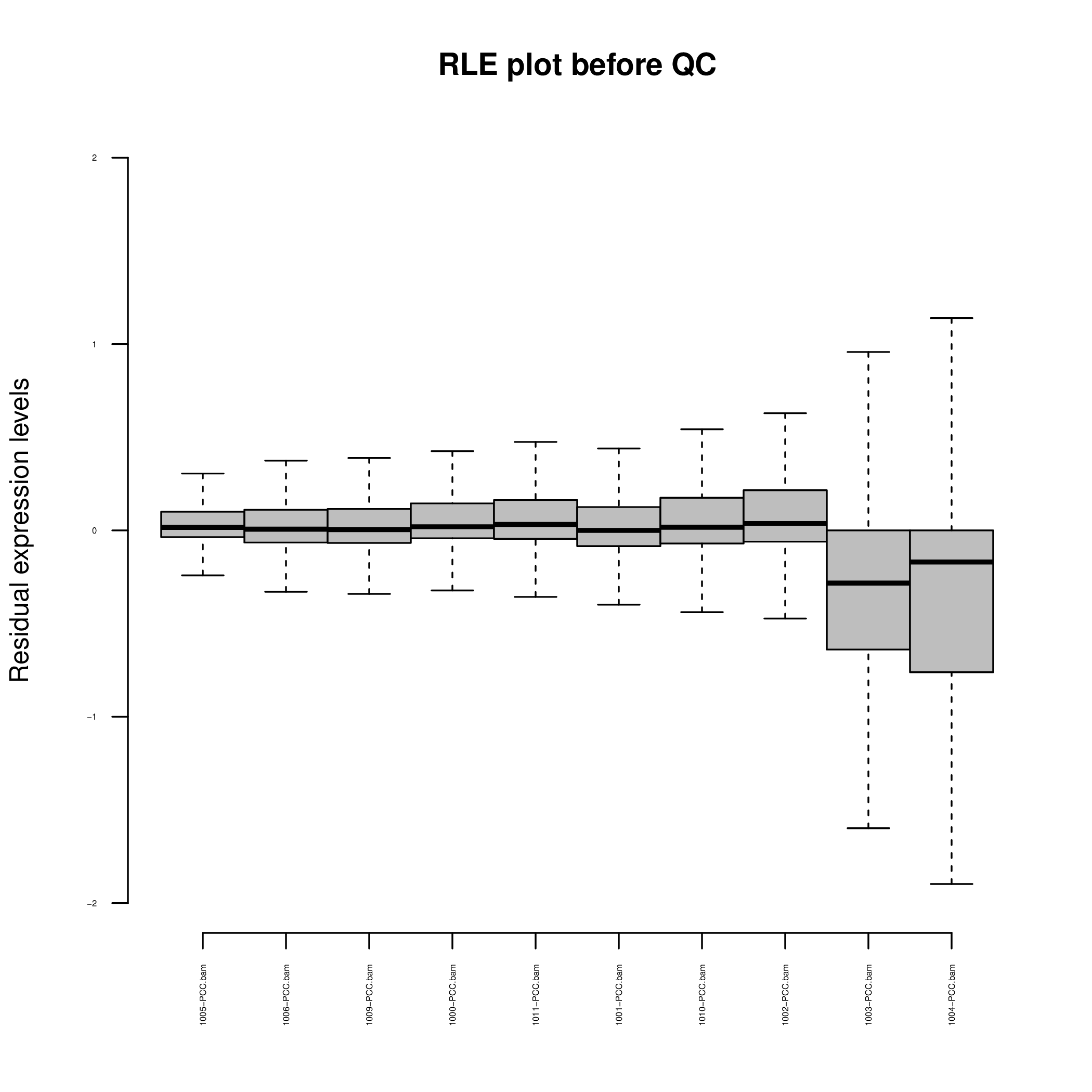
Figure 1B. Bulk RNA-Seq Quality Control Relative Log Expression Residuals.
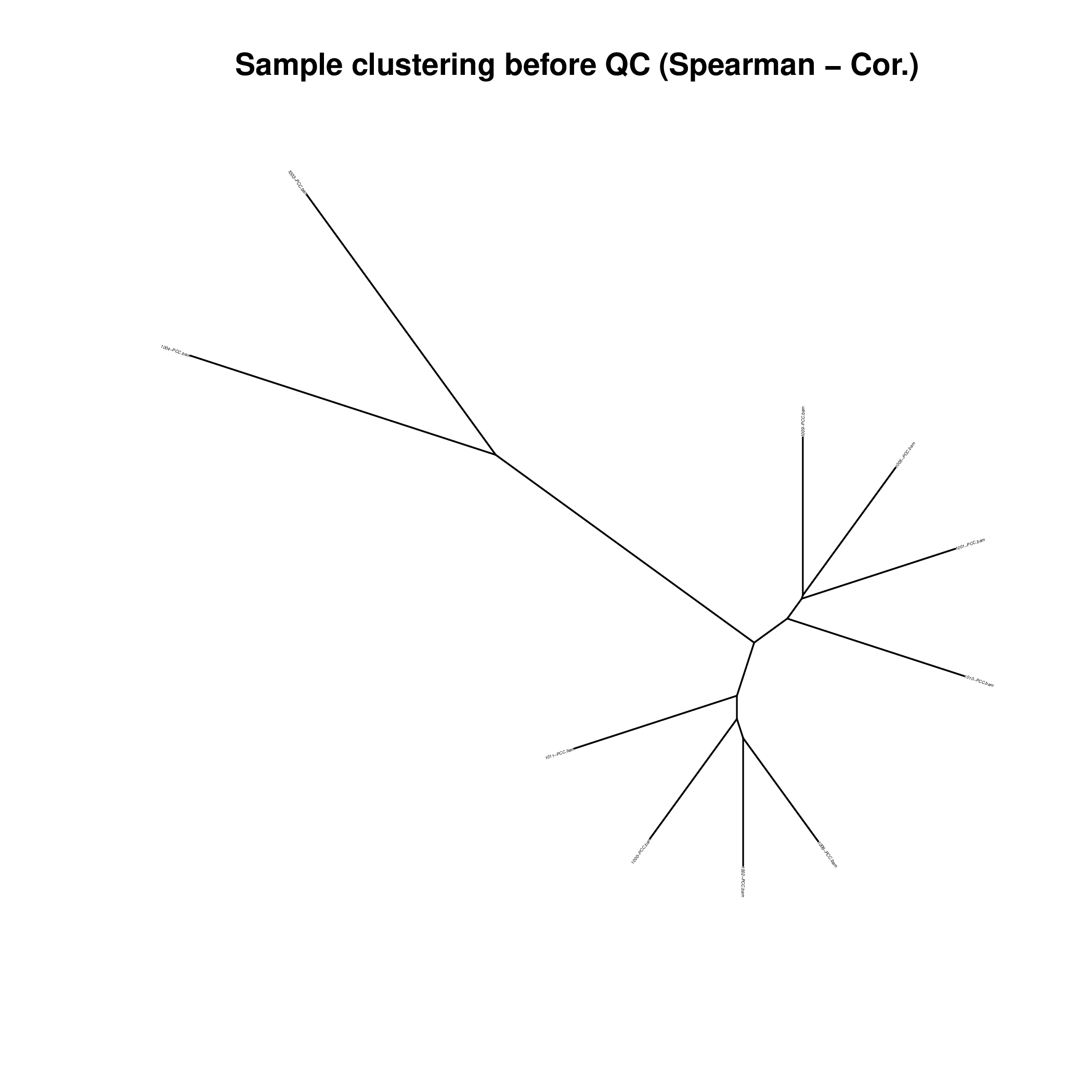
Figure 1C. Bulk RNA-Seq Quality Control Mahalanobis Distance P-Value Clustering.